
記住我
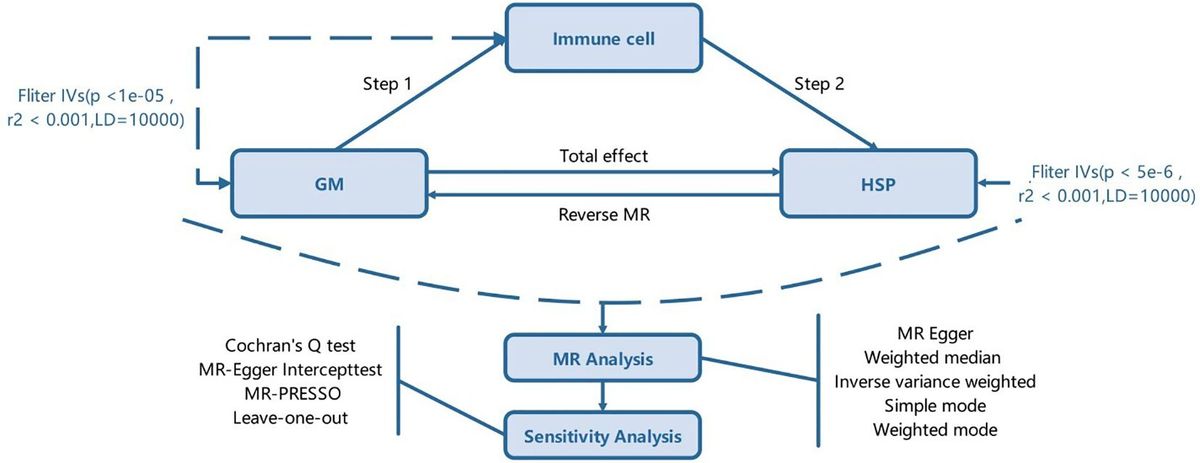
One and half decades after the ‘immuno-oncology tsunami’ that hit the clinical development landscape and shifted the momentum of treatment modalities, multiple new immunotherapy agents are now on the horizon. This progress raised optimism for many late-stage cancers for which treatment options are heavily exhausted. However, a second breakthrough remains elusive. The overall response rate (ORR) also remains static (20–40%) for all cancers. A rapid surge of therapeutic targets for developing new checkpoint blockades and other immune-agonist classes is on the ground (1, 2). There is also a new wave of developing RNA-based cancer vaccines to provide deep, durable memory responses (3). One vaccine candidate in recent time showed encouraging outcomes in lethal pancreatic adenocarcinoma (4). Aligned with this momentum, we witness an explosion of combination trials that aim to enable better survival outcomes (5, 6).
Colorectal cancer (CRC) is considered a global epidemic; over the decades, it has emerged as a pivotal cancer type affecting the younger population and is associated with late-stage detection and poor overall survival (OS) (7). CRC individuals with DNA mismatch repair deficiency (MMRd) positively respond to immune checkpoint blockade (ICB). A striking response failure in mismatch repair-proficient (MMRp) patients shows the diminishing return of the same therapy. In CRC, most of the success stories for ICB, thus far, have been limited only to tumors that manifest MMRd. However, this subtype is represented by only 15% of all CRCs and is far behind endometrial cancers (30%) and gastric cancers (20%) (8). For MMRp, ORR is not very different (10–15%) between TMB high and low CRC (9, 10). This realization prompted looking for effective treatment options for the MMRp subtype. The trend implies the necessity of addressing unmet needs both at personalized and population levels. At the heart of this challenge is the underlying complexity of the tumor microenvironment and its unpredictable dynamic immune milieu that form a barrier to effective therapy (11).
In this review, we discuss the changing clinical landscape of MMRp-dependent cancer indications (mainly CRC) and their uniquely hostile tumor microenvironment that hinders the success of current immune-based interventions. Both conceptual progress and clinical translation are illustrated in the light of rapidly evolving spatial biology contexts like tertiary lymphoid structures and gut microbiomes. We discuss the perspectives and challenges of biomarker-guided treatment selections for MMRp agonist cancers. We also highlight the alternatively actionable DNA repair pathways as emerging vulnerabilities to combat the treatment dilemma. Finally, we presented significant progress on the horizon of patient-derived functional ex vivo platforms that raised the hope of bridging the critical mechanistic gaps between drug pipelines and informed clinical decisions.
2 Molecular alterations defining MMRp evolutionary trajectoryA pan-genomic analysis from the 100000 Genome Cancer Program integrating genomic and clinical data revealed the highest enrichment of specific DNA. It deciphered MMR signatures in MSI-high (i.e. MMRd) colon adenocarcinoma and uterine corpus endometrial carcinoma. MMRp in this spectrum showed a negative association with survival compared to MMRd patients. Germline variants of MMR in this study found their link with the onset of colon adenocarcinoma at an early age (12).
2.1 Facets of intratumor heterogeneityThe intertwining intratumor heterogeneity (ITH) with TMB and TILs contextually represents a complex biology. ITH, either primary, adaptive or acquired during treatments, is considered a spatiotemporal bottleneck for a high response rate and duration of response. ITH encompasses genetic, phenotypic and dynamic tumor microenvironmental milieu and orchestrates therapy resistance. It also leads to the evolution of new resistant clones or the expansion of drug-tolerant persisters (13). Deciphering this ITH through the lens of MMRp and developing strategies to combat ITH mutations in tolerant cells are vital for adopting rational intervention. The study in the autochthonous mice model of lung and colon cancers highlighted that high TMB and MMRd do not guarantee immunogenic tumor infiltrating lymphocytes (TILs) and a positive response to checkpoint blockade. The subclonal escape of T cell response in these tumors was orchestrated by an immune-mediated increase in clonal diversity (14).
Instead of relying explicitly on genomics, the gradient of TME modulators like chemokines, neoangiogenesis, blood vessels, nutrients, and oxygen, along with ECM stiffness in time and space, play pivotal roles (15–17). Echoing this realization, the immune milieu is thought to collectively provide an actionable dynamic niche that interacts with the drugs and make the tumors reactive to ICB (18, 19). CRC show properties of reversible (mutation-independent) drug tolerance where recurrence is imminent after tumor cells are relieved from therapy pressure. Interestingly, barcoding and mathematical modeling suggested that equipotent clonal complexity is maintained for all cells throughout this process without any temporospatial loss. Under such conditions, tumors mimic a developmentally programmed diapause state at transcriptomic and signaling levels to overcome environmental turbulence (20). Indeed, drug-tolerant and disseminating tumor cells are, in general, notorious immune evaders that take advantage of being unnoticed by the immune radar to escape the primary sites and survive as silent perpetrators (21, 22).
2.2 MMRp clonal heterogeneity: more than a binary classAlthough MMRp and MMRd are binary molecular classes, recent profiling identified an intermediate category, i.e. heterogenous MMR or MMRh. The clonal overlap of MMRp and MMRd distinguishes it from the two classical subtypes. Gene expression analysis of CRC identified 14.5% of MMRd and 4.5% of MMRp cases as shared with this MMRh. The MMRh subclass allegedly evolves from double MMR gene loss. It is mechanistically linked to high TMB, TILs, and CD8 exhaustion phenotypes. High TMB (70 mut/mb) is attributable to higher subclonal variants. Genes associated with the MAPK pathway, antigen presentation and IFN-γ signaling pathway were significantly upregulated in MMRh class compared to MMRp (23). Moreover, 6-thioguanine and TMZ-induced enrichment of MMRd clones in MMRp tumors yielded encouraging outcomes. In two isogenic mice CT26 cell lines of MMRp (Mlh1+/+) and MMRd (Mlh1-/-) backgrounds, cross-complementing MMRp tumors selectively with MMRd clones rescued the immune surveillance program. MMRp clones, challenged with at least 50% MMRd cells, elicited tumor rejection. Both chemical induction and clonal competition strategies were able to underpin a heterogeneous MMR context of improved anti-tumor immune reactivity (24). This study in mice cell lines of CT-26 with MMRp backbone affirmed that reconstitution of MMRp clones with MMRd powered them to eliminate the MMRp fraction. The clonal and sub-clonal contexts of these two studies highlighted the differences in experimental approaches and interpretations of results. Specific TMB/neoantigens low subclones of mice tumors can evade an immune attack due to defects in cross-priming or active interference by dysfunctional T cells or immune ignorance. Moreover, these tumors are thought to acquire immunogenicity during in vivo repropagation at the clonal level but not at the sub-clonal level. As a result, the rapid contraction of MMRp clones was attainable (25, 26). This divergent clonal journey revealed the dynamicity of the ecological and evolutionary landscape of MMRp (27).
2.3 Altered gene regulation and mutations in MMRpFurther dissection of MMR status at the molecular level sheds light on the key regulatory elements driving transcriptomic machinery. Mutations (indels) in diverse CRC samples revealed that MSI-high CRC largely harbor gained enhancers that selectively offer the privilege of recurrent growth of these tumors through increased affinity for putative transcription factor, e.g. Forkhead Box D4 (FOXD4) and target gene overexpression that is regulated by these enhancers (28). Some of these genes have been implicated in chemoresistance, unrestricted oncogenic EGFR signaling, regulation of proliferation and apoptosis in primary CRC tumors and in established MSI cell lines. In the MSS cohort, the occurrence of enhancer indels was found to be at a much lower rate. Compared to MSS, MSI-high CRC has shown 50% more gained enhancers at TGTTT(Tn). It was linked to H3K27ac enrichment. A panel of 10 different FOX- transcription factors (FOX-TFs), encompassing FOXP2, FOXC1, FOXD3, FOXM1, FOXJ3, FOXA1, FOXO1, FOXO3, FOXG1 and FOXA2, presented the consensus sequence, a signature motif at indel alleles, and confirmed the binding affinity of FOXs. However, due to the degenerative nature of this consensus motif, findings did not specify the dominance of any single FOX member from the family in the enhancer activation. Instead, it proposed additional studies to fill this gap, elucidate cooperative interaction with other factors and inter-tumor heterogeneity, if any (29). This also implies the need for further delineation of other enhancers and super-enhancers to gauge their differential impact on the oncogenic driver alterations, making MMRd and MMRp tumors more vulnerable to therapies (30).
About 30% CRC are hereditary and germline predisposition affects CRC susceptibility. About 5%–7% of CRC cases are caused by germline mutations. Classic hereditary CRC syndromes are mainly due to germline mutations in APC, MUTYH, and mutations in genes encoding four mismatch repair enzymes, namely MSH2, MSH6, PMS2 and MLH1 (31, 32). Pathogenic somatic mutations (predominantly biallelic) in coding regions of one of these four mismatch repair enzymes lead to the development of MMR deficiency of CRC and eventually give rise to an MSI phenotype. Interestingly, Lynch syndrome (LS) or hereditary nonpolyposis, CRC, represents one-third of these MMRd, is an early onset CRC, therefore suggesting precedence of germline mutations (33). Similarly, tracking the germline defects in the MMRp genes provided significant screening opportunities for CRC of hereditary background. DNA mismatch repair protein O6-methylguanine DNA methyltransferase (MGMT) is frequently detected in CRC. Its epigenetic inactivation in somatic clusters is prevalent. However, elucidating whether the same defects are perpetuated in the germline background, particularly in MMRp, showed non-confirmatory results. No promoter methylation linked to constitutive MGMT inactivation was confirmed. Indeed, two rare heterozygous germline variants were detected in 4 families. Further segregation of these variants in neoplastic lesions in the affected family suggested that more data are needed to establish their link to MMRp in familial CRC (34). Although most CRCs by default are MSS, and MMRp represents low TMB, the study showed that 7.5% of colon and 9.5% of rectal cancers of this background also had high TMB (defined by >10 mutations per Mb). Interestingly, KRAS mutations and gene mutations involved in DNA damage repair (DDR) machinery and epigenetic modifiers were high in TMB-high MSS tumors. These findings suggest that molecular alterations are potential triggers of TMB in CRC (35). Like MMRh, these subsets open new opportunities for target dependency and vulnerabilities to gauge their promise in differentiated intervention.
3 Mechanistic constraints of MMRp tumors and strategies reversing themA hostile MMRp context of tumor-immune microenvironments (TiME) is a formidable therapeutic challenge in achieving clinical success. Low TMB and immunological ignorance are two critical hallmarks of the MMRp tumor ecosystem. Further complexity of a tangled network emanating from dysbiosis of microbiome and problematic metabolomics has the propensity to escalate the suppressiveness of the microenvironment to diverse therapy regimens. Understanding the dynamicity and dimensionality of this misdirected microenvironment and its immune restrictive milieu is pivotal to developing novel rationale interventions that can mount an all-out attack on these tumors (Figure 1).
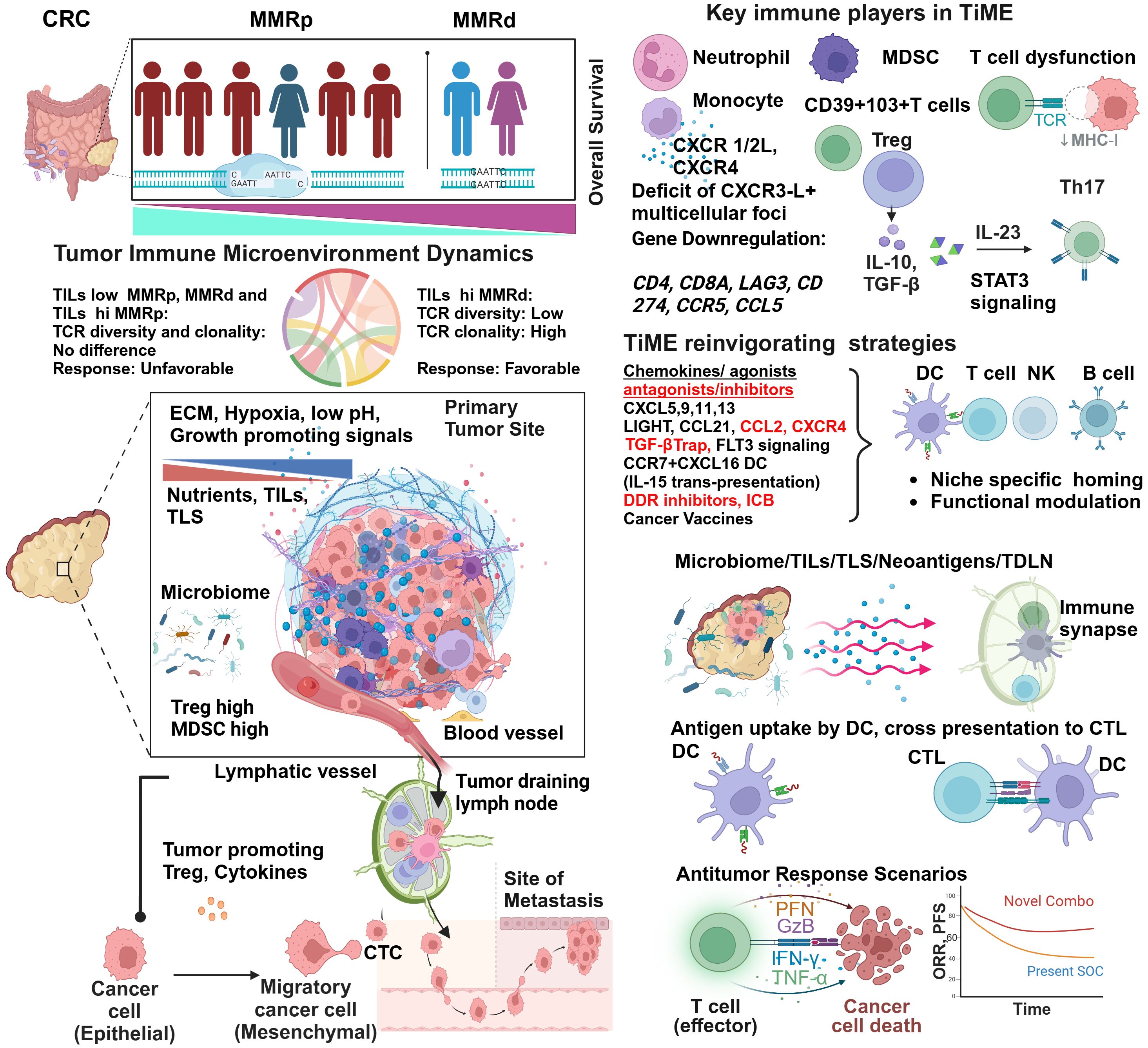
Figure 1 Tumor immune microenvironment of MMRp displays mechanistic constrains: potential strategies of reinvigorating. Despite representing more than three-fourths of the entire CRC population, the majority of MMRp CRC belongs to high-risk and poor prognoses. The TiME of CRC has multiple mechanistic barriers that hamper the therapy success. High TCR diversity and low TILs density in the margin and core at primary CRC sites are associated with desmoplastic stroma, growth-promoting oncogenic signaling, poor blood vessel density and patterns leading to oxygen and nutrients deprivation. Poor microbiome context and active involvement of Treg and MDSC in a traditionally low TMB milieu critically orchestrate immune evasion of disseminating tumor cells to distant sites through unguarded blood vessels and immunologically skewed TDLN’s surveillance. The phenotypic analysis of TILs confirms the presence of immune cell types of suppressive functions and corresponding cytokines and chemokine networks that protect the tumor from immune attacks. Several agonists and antagonists of chemokines, TGF-β-targeted therapy, and vaccines can act in concert with other strategies to reinvigorate and stabilize TILs and TLS via niche-specific recruitments of the anti-tumor immune army. Finally, augmenting neoantigen load and DC functionalities cross prime CD8+T cells. In totality, other immune and non-immune targets present in the TiME provide an opportunity to rationally target this challenging microenvironment in clinical settings and improve the response.
3.1 TiME perspectives and TILs in MMRpOne critical differentiator in response to ICB between MMRd and MMRp is TILs density and its proximity (core and invasive margin) to target tumors. While a higher number of MMRp CRC patients are ascribed to be TILs deprived, i.e. they have lower TILs density, inter-tumor TILs heterogeneity (30–90%) in MMRd is not uncommon (Figure 1). This contexture includes a spatial heterogeneity of CD3+ and CD8+ TILs in the invasive margin and tumor core. Interestingly, a diminished TILs footprint in MMRd acts to underpin MMRp+ CRC response to ICB, a feature paralleling their functional TiME orientation (36).
3.1.1 TCR diversity and clonal expansionIt is intriguing to note that not only the TILs density and distribution but TCR repertoire and clonal landscape in MMRp CRC are also different from its MMRd counterpart. While T cell clonality and the richness of TCR repertoire have similar imprints in MMRp with TILs low and TILs high context, a sharp contrast contradicts this signature in MMRd tumors. In MMRd, higher T cell clonality was observed to be matched with lower TCR richness in TILs high tumors while comparing them with TILs low tumors within the same MMR class (Figure 1). It is imperative to note that under high TMB, T cells are clonally expanded in MMRd with high TILs. The constraints of low TMB in MMRp made both TILs low and TILs high tumor uniform in their clonal expansion program, and they maintained similar TCR diversity (37). These findings show that T cell clonal dynamics in TMB with a low and high background may reciprocally impact immunosurveillance. Studies in other cancers claimed that global TMB alone is not a perfect proxy for the foreignness of antigens. An evolutionarily persistent TMB, due to single or multiple copy regions per cell, provides a better response opportunity by creating a bottleneck for tumors that is difficult for them to overcome (38).
3.1.2 TILs numbers and spatial contextureThe organ specific immune atlas is emerging as an important platform to capture deeper phenotypic perspectives and insights. Single-cell RNA sequencing revealed the existence of a distinct immune hub in a spatially defined CRC cancer-immune network. A shared myeloid-rich inflammatory immune hub in tumors below the colonic lumen and CXCR3-ligand positive anti-tumor multicellular foci accompanied by activated T cells in MMRd tumors contextually distinguish them from MMRp (39). Like spatial TILs, quantitative analysis of global TILs in MMRp is a logical step in defining their prognostic impact. In high power field (HPF) quantification of TILs, based on a threshold set as >3 (high) vs <3 (low), five years of recurrence-free survival was observed to be higher in MMRp CRC with high TILs (94.6%) compared to their low TILs counterpart (77.9%). More importantly, in multivariate analysis using stages and TILs as key discriminators along with MMRp status, the higher stage with high TILs resulted in a similar relapse-free survival (RFS) to that of the lower stage alone without impacting OS (40).
3.2 TiME beyond TILs: APC defects, γδT and NK cellsReactive TILs are not the only immune subset that encounters tumors. Multiple evasion points that perturb the TiME are contextually intertwined.
3.2.1 PD1+γδT cells in B2M defective immune networkIn the case of conventional antigen-presenting cells (APC), gene defects mainly govern β2 microglobulins (B2M) inactivation and HLA class 1 dysregulation in CRC. ICB under these circumstances increases γδT cell subsets with PD1, killer-cell immunoglobulin-like receptors (KIR) and cytotoxic activation markers in TME. These players, in concerts, determine the preferential retention of ICB responsiveness in cell lines of MMRd and MMRp backgrounds as well as patient-derived organoids that are defective of B2M gene and show concomitant loss of HLA class1 presentation machinery. HT-29 CRC cell lines of MMRp lineage retain B2M function compared to MMRd lines (HCT-15 and Lovo), where B2M gene defects (HLA -1 antigen presentation loss) in HT-15 cell lines instigate the most profound ICB response orchestrated mainly by PD1+ γδT cells in a coculture based drug reactivity assay (measuring Caspase3/7). The reintroduction of B2M genes in MMRd cell lines resulted in a loss of tumor killing by γδT cells in response to ICB under similar conditions. Further delineation of MMRd clinical CRC samples by multiplex spatial immune profiling suggested a remarkable increase in γδT cells in B2M defective cases. MMRd Patient-derived organoids with B2M loss elicited a better response by PD1+ restricted γδT cells (41).
3.2.2 FLT3L signaling defects in DC functionalityPreclinical models of MMRp CRC (accounting for 95% of all mCRC) revealed that these tumors preferentially spread to the liver following orthotopic implantation but are restricted when heterotopically implanted in a subcutaneous site known for its context deficit and poor vascularization. This complementary liver metastasis model importantly recapitulated the paucity of CD8 and DC, consequently maintaining the non-responsiveness to immune checkpoint blockade (ICB). Combined treatment of Feline McDonough sarcoma (FMS)-like tyrosine kinase 3 ligand (Flt3L) plus ICB therapy improved survival by enhancing dendritic cell infiltration (42) (Figure 1). Indeed, whole-genome analyses of metastatic colorectal cancers from a pan-cancer Hartwig database of 2256 MMRp samples confirmed that only 1.6% of these samples clonally showed B2M and concurrent loss of heterogenicity (LOH), limiting their prognostication impact (43). In another study, pexidartinib, a CSF-1R–directed tyrosine kinase inhibitor (TKI), in combination with durvalumab (anti–PDL-1) in CRC and other cancers, resulted in limited efficacy. Pexidartinib impaired the development and functionalities of DCs due to the inhibition of FLT3 signaling ex vivo and in vivo (44). These findings illustrate the importance of maintaining active FLT3 signaling to achieve reasonable response (Figure 1).
3.2.3 NK cells rescue B2M-driven immune dysfunctionInterestingly, in MMRd CRC, B2M mutations that canonically disrupt antigen presentation machinery showed paradoxical outcomes. It prevented disease recurrence, metastasis and helped manage prolonged survival. In this case, NK cell mediated inhibitory effects in the absence of HLA 1 defect prevented metastatic spread (45, 46). Unlike CRC, in endometrial cancer, defects in T cell activation signaling due to JAK1 mutation turn the tumors immune inert (47). Phase I/II multicenter study of autologous DC with Avelumab in mCRC for pharmacodynamics (pD), safety and efficacy showed well-tolerated outcomes but a modest 6-month PFS (only for 11% of patients). Interestingly, the rewiring of lipid metabolism against glutamine and glucose utilization and the generating reactive oxygen species (ROS) in response to this combination contributed to longitudinal progression. There is an urgent call for tailoring novel therapies to target this dependency as a vulnerable checkpoint (48).
3.3 Tertiary lymphoid structure in MMRp is an elusive immune hubIn recent years, conceptual progress and clinical promises of tertiary lymphoid structures (TLS), a specialized tumor-immune microenvironmental niche, have attracted attention (49). They have a concerted influence on priming/amplification//licensing itineraries in TiME. The TLS army involves diverse lineage-specific subsets like plasma cells/B cells, different DCs like conventional DC (cDC), follicular DC (fDC) and other myeloid and lymphoid-derived cell types. In CRC, TIL, TLS and their abundance are mainly elucidated in MMRd (50). For MMRp, the ongoing efforts dissect niche-dependent immune evasion and design rationale therapies that can enrich the TLS footprint and redefine ICB response. In parallel, microbiome-immune crosstalk in eliciting anti-tumor response is appreciated in CRC and implicated the roles of TLS (51). From the qualitative and quantitative perspectives, the size, composition and spatiotemporal dynamics of TLS and non-TLS immune hubs like TILs and lymphonets promise new therapeutic modalities (52). A 56-marker multiplex IHC-driven cellular classifier (CODEX) at the invasive front identified CD4+PDL-1-positive cells in the granulocyte neighborhood as the only positive prognostic marker in high-risk advanced-stage CRC. In contrast, the lack of inter-compartment connectivity in TiME contributes to unfavorable outcomes (53). At preclinical levels, however, there are limitations to the potential human translation of TLS. One reason is that besides wide gaps in TME, mice tumors exhibit rapid and aggressive growth. This property inherently restricts the scope of mature TLS formation within a defined temporal neogenesis window. The cells that populate an immature or suppressive TLS, e.g., Breg, Treg and MDSC, can also perpetuate in the MMRp (54–56).
3.4 Chemokines and immunomodulators in homing and reinvigoration of MMRpChemokines, released by tumor cells and other cell types like stromal fibroblasts and endothelial cells, act as chemo-attractants. Through engaging cognate receptors, they recruit immune cells that are anti-tumor or immune suppressive (pro-tumor) in functions. Multiple interactive chemokine axes also influence therapy outcomes. These depend on the types of chemokine ligands, cognate receptors on the target cells and specific TME contexts (57, 58).
Mechanistically, the reconstituted chemokines milieu could be a critical orchestrator of reinvigorating the depressed TLS and TiME. Spatially delineated biomarkers or ‘biopatterns’ shed light on the niche-specific recruitment and interactions of immune cells in TME. A comprehensive knowledge of their contexts is important for microenvironment-guided therapy selection (58, 59). The MMRp tumor-immune niche is the home of several suppressive immune subsets. Monocytes, neutrophils, MDSC, Tregs and Th17 cells are lead players in this domain and are responsible for maintaining a tumor-friendly suppressive network (Figure 1 and Table 1).
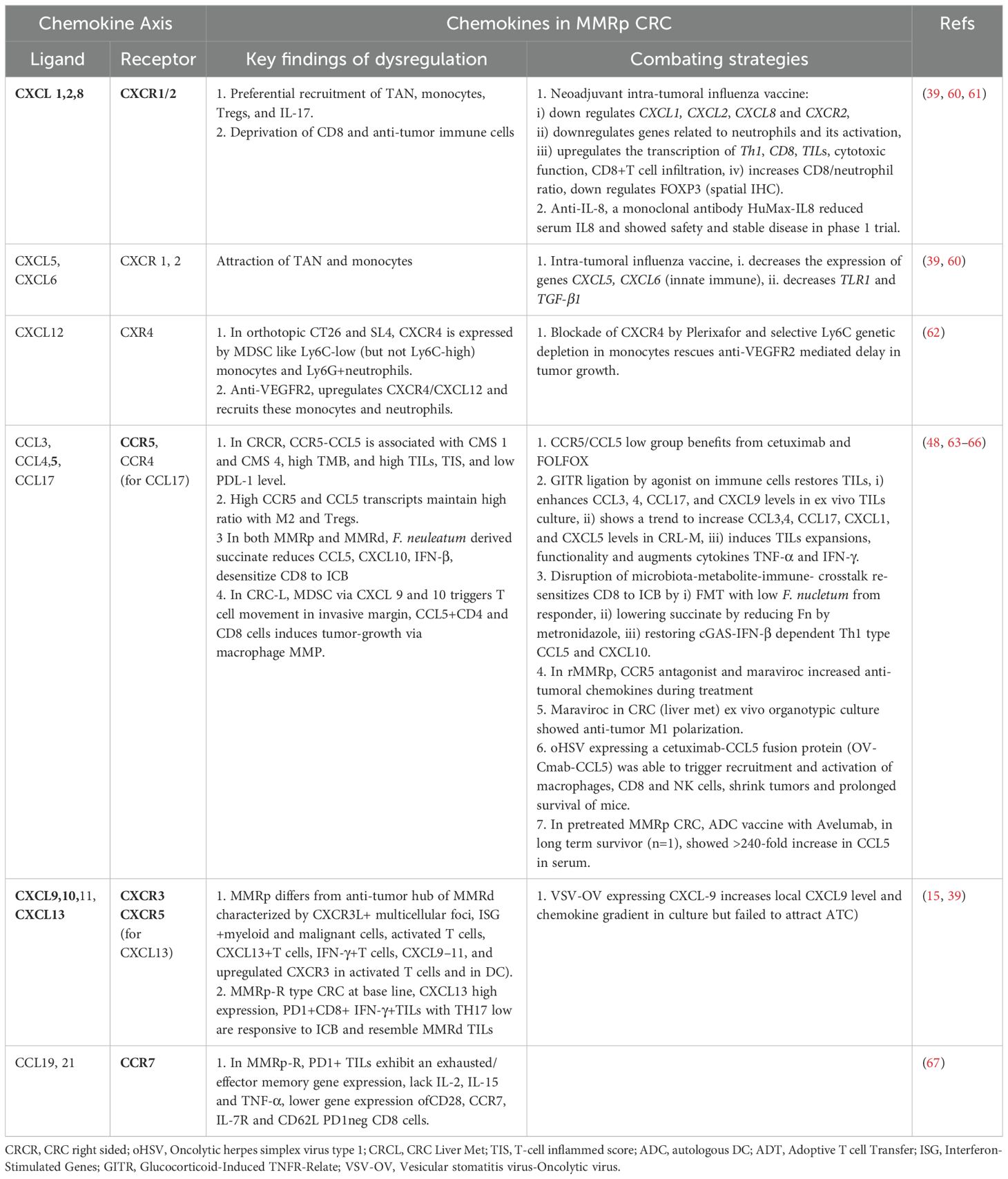
Table 1 Key Chemokines, their receptors, functions and combating strategies in MMRp CRC.
3.4.1 CXCR1/2, TGF-β signaling and Th17Preferential recruitments of tumor-associated neutrophils (TAN), monocytes, other myeloid- derived suppressor cells (MDSCs), Tregs, and Th17 functionally cooperate through chemokines like CXCR1/2 and CXCR4-CXCL12. In this network, IL-10, TGF-β, IL-10, IL-23, and STAT3 signaling worsen the prognosis (58, 67–70). Their interplay hampers the prospects of therapies. For example, anti-VEGFR treatment in MMRp mice orthotopic model triggers a positive feedback loop that further upregulates CXCR4-CCL12 and recruit monocytes and neutrophils in the TME (62).
3.4.2 Critical regulation dynamics of CCR5-CCL5 axisIn a hyperpolarized TME, CCR5 and CCL5 in MMRp are tightly associated with the right-side colon, poor prognosis-related consensus molecular subtypes 1 and 4 (CMS 1 and CMS 4), high TMB, and high TILs. Different myeloid and lymphoid subsets like M1, M2 macrophages, B cells, CD4, CD8, T regs and NK cells, in coordination with PDL-1, CTLA-4 and PARP, orchestrate a depressed immune network. It is evident that CCR5/CCL5 low group benefits from targeted therapy of cetuximab and FOLFOX (71). Paradoxically, past research showed a poor prognostic link to CCR5. The study showed a marginal improvement in specific combinations. However, due to the unavailability of data from retrospective analysis, confounding effects cannot be ruled out (71). Collectively, these modalities can guide the homing of immune cells in MMRp tumors known for traditionally lacking reactive TLS footprints. As illustrated in Figure 1 and Table 1, TiME of MMRp selective tumors has deficits in TILs and TLS. It shows a distinct bias for Treg and MDSC (mainly monocytes and TAN). Moreover, the immunosuppressive cytokines that MDSCs augment, facilitate epithelial-mesenchymal transition (EMT), increase the propensity for distant metastasis of disseminated tumor cells and confer failure to therapies (67, 72, 73). In the invasive margin of CRC liver metastasis (CRC-L), CCL5+CD4 and CD8 cells, recruited by myeloid-derived CXCL9 and CXCL10, attract macrophages promoting invasion and tumor growth by MMP (65). However, the same CCL5 in T cells is impaired under succinate high microenvironment created by F. nucleatum and mediate ICB nonresponse (66). These findings suggest the contrasting roles of CCL5-CCR5 axis in different TME contexts.
3.4.3 Chemokine agonists and antagonists in reshaping MMRp TiMEA number of therapeutic options are emerging on this horizon to fill the vacuum. The key modulators are chemokine agonists and antagonists that reciprocally orchestrate niche-specific recruitment and modulation dynamics. Functionally active chemokine axes, their dysregulation in MMRp and mechanistic interventions are illustrated in Table 1. A rational approach in MMRp can unlock the potential of reinvigorating its TiME. Retaining or reconstructing niche specific chemokine networks led by A) CXCR3 and its ligands CXCL9, 10, 11, CXCR5 and its ligand CXCL13, B) CXCR2 and its ligand CXCL5,6, C) CCR4 and its ligand CCL17, and D: CCR5 and its ligands CCL5, and additionally CCR7 hold promise in this space (reviewed in 57,58). Chemokines like CCL1, CCL2, CCL8, CCL12, and their receptors (e.g. CXCR1/2, CXCR4) reciprocally facilitate the homing of myeloid suppressors (monocytes, TAN, MDSC) and Treg. The drugs or antagonists targeting their actions can rejuvenate the immune reactive interface (58, 65, 73).
Neoadjuvant intra-tumoral influenza vaccine in MMRp CRC showed downregulation of pro-tumor chemokine genes, TGF-β genes. It concomitantly upregulated genes involved in Th1, CD8, increased TILs and cytotoxic function. The same vaccine decreased the Treg transcription factor FOXP3 at the protein level (60). Autologous dendritic cell (ADC) vaccine with Avelumab showed a decline in serum CCL2 level in pretreated MMRp CRC and a 240-fold increase of serum CCL5 in a long-term survivor (48). Inhibition of CXCR4 by Plerixafor or selective Ly6C targeted genetic ablation in monocytes rescues mice from anti-VEGFR2 induced tumor progression (62). CCR5 antagonist maraviroc has been tested in preclinical ex vivo tumor culture. In an independent clinical study, it demonstrated anti-tumor macrophage repolarization and anti-tumor chemokine augmentation, respectively (65, 74). CXCR3/5 ligands selectively recruit cells like DC, CD8 and T helper 1 as part of a niche-specific homing program (39, 58, 75). Therapies rely on agonists that drive the targeted enrichment of chemokines like CXCL9, 10, 11 and 13 and facilitate CD8 recruitment. For example, Glucocorticoid-Induced TNFR-Related (GITR) ligation by its agonist enhanced CCL3, CCL4, CCL17, and CXCL9 levels in CRC derived TILs in ex vivo culture. It induced TILs expansions, functionality and augmented proinflammatory cytokine (e.g. TNF-α, IFN-γ) (64). Gut microbiota adds a layer of criticality to this interface. Disruption of microbiota-metabolite-immune- crosstalk with low F. nucleatum (Fn) from responder or reducing F. nucleatum by metronidazole diminished the local succinic acid in TME and re-sensitized CD8 to ICB. This intervention also restored cGAS-IFN-β dependent CCL5 and CXCL10 following their decrease by high succinate (66). Anti-IL-8 antibody reduced serum IL-8 in phase 1 trial (61). Oncolytic virus expressing CXCL-9 restored local chemokine gradient but failed to recruit adoptive T cells (ATC) in culture (15).
Chemokines function as important modulators of TLS in both MMRd, MMRp scenarios (76, 77). Chemokines like LIGHT, LTa, CCL21, and APC activating agonists for TLR4 and CD40 are critical druggable targets to boost TLS (78). CCR7+ CXCL16+DC mediated trans-presentation of IL-15 to effector-like CTLs in perivascular niches orchestrate their survival and expansion. This survival and proliferation signaling loop averts an irreversible terminal differentiation of CTLs into the hypofunctional or tolerant state and maximizes the quality of response (79), (Figure 1 and Table 1).
IL-15 trans-presentation, TGF-β-Trap with anti-EGFR, DDR inhibitors and cancer vaccines are also under active development to overcome the outstanding challenges (48, 60, 79–81). While mechanistically compatible TILs in such scenarios may provide a milieu for immune-based interventions, other tumor intrinsic evasion strategies can still be a barrier that avert T cell-mediated attack of tumors (22). For example, perforins and granzymes are two critical polarized cytotoxic effector molecules released from activated NK and T cells. Perforins act as a port of entry for granzymes. However, tumor cells manipulate their inherent ESCRT-mediated membrane trafficking strategy to repair these pores and, therefore, block the entry of granzyme (82).
3.5 Lymph node niche and immune surveillance: TGF-β signaling interventionAt the systemic level, a compromised immune activation network signals a prospective disease that is often advanced beyond primary sites. A recent study also proposed that preserving the tumor-draining lymph nodes (TDLN) may benefit anti-tumor immune reactions. Based on CRC data, dissection of immune phenotypic profiling showed differentiated TILs and TCR repertoire dynamics in lymph nodes (LN). This profiling separated MMRd from MMRp. In general, lymph node lymphocytes (LNL) show an intermediate functional state when compared with peripheral blood (lowest) and intratumor TILs (richest in tumor-reactive TILs). Stage-dependent TIL analysis also showed higher TILs in early-stage MMRd compared to matched early-stage or late-stage MMRp. Cytotoxicity-related genes also maintain similar enrichment patterns in MSI-H/MMRd cohorts. In this continuum, shared TCRs analysis of TILs showed their lower percentage in the proximal LN (pLN) of MMRp compared to MMRd. These data show the potential benefit of avoiding excessive non-metastasis LN dissection in MMRd (83, 84). CRC from MMRp origin contains neoantigen reactive autologous TILs co-expressing CD39 + 103+ T cell subsets in the CMS4 (less immunogenic) context. These T cells are known for promoting a paracrine TGF-β signaling loop and have the worst prognosis. Further delineation of checkpoint status targeting TGF-β and its trap with PDL-1, in this context, expected to reinvigorate TIL effector functions (69, 85, 86). However, an anti-PD-L1:TGF-β trap fusion protein directly targeting MSS-positive metastatic CRC failed to control the recurrence of ctDNA and, instead, elevated the level of ctDNA (86). Other strategies of dual targeting TGF-β with EGFR (e.g. BCA 101) are under development. Its combination with ICB in preclinical in vitro coculture assay using PBMC in EGFR-insensitive human colon cancer cell line HCT-116 (MSI hi) showed synergy with a high TGF-β footprint. Immune-reconstituted human colon cancer HT-29 (MSS) in mice xenograft model mechanistically elicited a potent immune-mediated anti-tumor response upon BCA 101 exposure (80) and Figure 1. More studies intersecting the TGF-β crosstalk in suppressive MMRp are needed to boost the quality of responses. KRAS mutant CRC and similar cancers were portrayed as undruggable until recently. A lymph node-targeted KRAS mutant peptide vaccine with a CpG oligo adjuvant (Amph-CpG-7909) in the AMPIFY-201 trial tested this therapy on 5 CRC patients, all from MMRp background. In 84% of cases, it showed T cell response ex vivo. Of this response, 54% involved both CD4 and CD8-specific T cells. In 84% of cases, there was a decline in biomarker (ct-DNA level) from the baseline, and in 24% (3 Ca-Pancreas and 3 CRC) cases, total clearance of biomarkers was achieved (87) and Figure 1. One outstanding question is what should be an ideal therapy plan for CRC patients with preexisting autoimmune conditions. IL-17-IL-23 axis is a clinical target in multiple autoimmune disorders (88). Therefore, a rational combination of these agents in MMRp patients with existing and new I-O and non-IO agents deserves evaluation through proper trials.
4 Onco-microbiome and metabolome interface in MMRp tumorsAmong different theories, a complex interplay of intrinsic and extrinsic factors and cellular plasticity governs the initiation, maintenance and progression of cancers (89). The cancer risk mapping in higher mammals identified new attributes independent of body size and age that were thought to accommodate more cancer-causing mutations or “bad luck” mutations, initially coined by Tomasetti and Vogelstein (90). These risks include diets and loss of the gut microbiome homeostasis (91). New insights highlighted the clouds of complex systemic landscape in the frontiers of cancer hallmark. Besides metabolic alteration, ageing and obesity, tissue macro and microenvironment, myeloid dysfunction and other physiological dysregulation, genetic and environmental factors, this conceptual progress reiterates microbiome as a lead dimension (92).
4.1 Tumor invading gut microbiota and its orientation in MMRpIn recent years, the role of the microbiome in redefining novel immune therapy has fascinated clinicians and researchers alike. Physiological decoding of the microbiome and its metabolite derivatives (e.g., amino acids and short-chain fatty acids) in MMRd and MMRp identified functionally distinct footprints. For example, enrichment of Bacteroides fragilis and sulfidogenic Fusobacterium nucleatum (Fn) were profound in MMRd. In contrast, Bacteroides fragilis was deprived in the MMRp tumor-microbiome interface. Both these species are linked to a maladapted metabolic landscape in the gut niche (93, 94). To further narrow down at the level of clades, a recent study identified that clade 2 of the Fn subspecies animalis (Fna) strain is predominant behind intra-tumoral loads following heavy colonization in the CRC niche (95). Fusobacterium nucleatum gets the upper hand in an MMRp ecosystem. It confers resistance to chemotherapy, promotes Wnt signaling, binds to TIGIT through its Fap2 component, and activates inhibitory cytokine-producing Treg and M2 macrophages. TLR4-NFkB signaling under such conditions is impaired in chemo-resistant CRC due to the upregulation of autophagy and anti-apoptotic signals (96, 97). Among different metabolites, bacteria-derived inosine acts via A2A adenosine receptor (A2AR) in Th1 cells, facilitates T cell and DC crosstalk and increases the metabolic fitness of CD8+T cells to trigger tumor killing (98). It serves as an alternative carbon source where there is a restriction of glucose availability to CD8 (99). The chronic inflammation due to a low-fibre diet and altered bacterial interaction with archaea insults the gut ecosystem, shifting a balance to dysbiosis (100, 101). This property also orchestrated a metabolomics bias where MMRd tumor had more association with host protective amino acid biosynthesis (102).
4.2 Interplay of the microbiome and immune niche: TGF-β and Th17 paradigmLoss of resistance to the colonization of harmful invading bacteria, a new hub created by them in the vicinity of the tumor and inside tumor core including immune cells challenge the host protective anti-tumor immune function. Th17 cells help maintain homeostasis (eubiosis) in a normal gut. However, damage caused by bacterial invasion on gut epithelia triggers the loss of IL-17RA. The systemic spread of Th17 cells and B cells to distal organs facilitates tumor promotion via Dual oxidase 2 (DUOX2). This study showed compartmentalized and context-dependent roles of IL-17 signaling (103). Specific cellular contexts of the IL-1 receptor (IL-1R) also determine the impact of microbial induced IL1 signaling on CRC pathogenesis. While IL-1R deletion in epithelial cells blocks CRC progression independent of inflammation, the same defects in the T cells and the myeloid cells (mainly neutrophil) restrict and exacerbate tumor growth and progression, respectively, following the microbial invasion in tumors (104). The targeted ablation of source bacteria can block the compensatory loop. Similarly, in the mice inflammation model, CD4-driven IL-10 production through macrophages augments IL-17 production (105). Fn is a dominant player in this paradigm. It drives a shift that augments formate production. Aryl hydrocarbon receptor (AhR) signaling promotes invasion and cancer stem cell properties in in vitro co-culture of Fn with CRC cells under this condition. In mice, Fn injection increases the Th17 cell expansion and tumor growth (106). Since IL-17 low MMRp tumors favor ICB outcome, similar cross-talk, and context are expected to persist in their suppressive TME. The γδT-17 cells represent another subset that drives an MDSC bias in CRC (67, 107, Figure 2).
4.3 Diversity and metabolites influencing MMRp milieuSystems-level diversity of the gut microbiome describes their influences in shaping CRC tumor niche. The 16S rRNA gene sequencing of dMMR (n=29) and pMMR(n=201) in tumors (T) and matched adjacent normal (N) tissues deciphered critical differences in their diversity both at alpha and beta levels. Overall, species diversity of gut microbiome (alpha diversity) was higher in the MMRd-T niche than in MMRp-T and MMRd-N. This comparative profiling showed significant differences (beta diversity) between MMRd and MMRp (Figure 2). Secondary Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis confirmed microbiota-related glycan metabolism, vitamins and nucleotide biosynthesis, active cell death, and defects in DNA repair machinery in MMRd tumors. Indeed, these properties favored PFS and OS upon immunomodulator exposure in MMRd and outperformed MMRp, where microbiota relies predominantly on lipid metabolism (108).
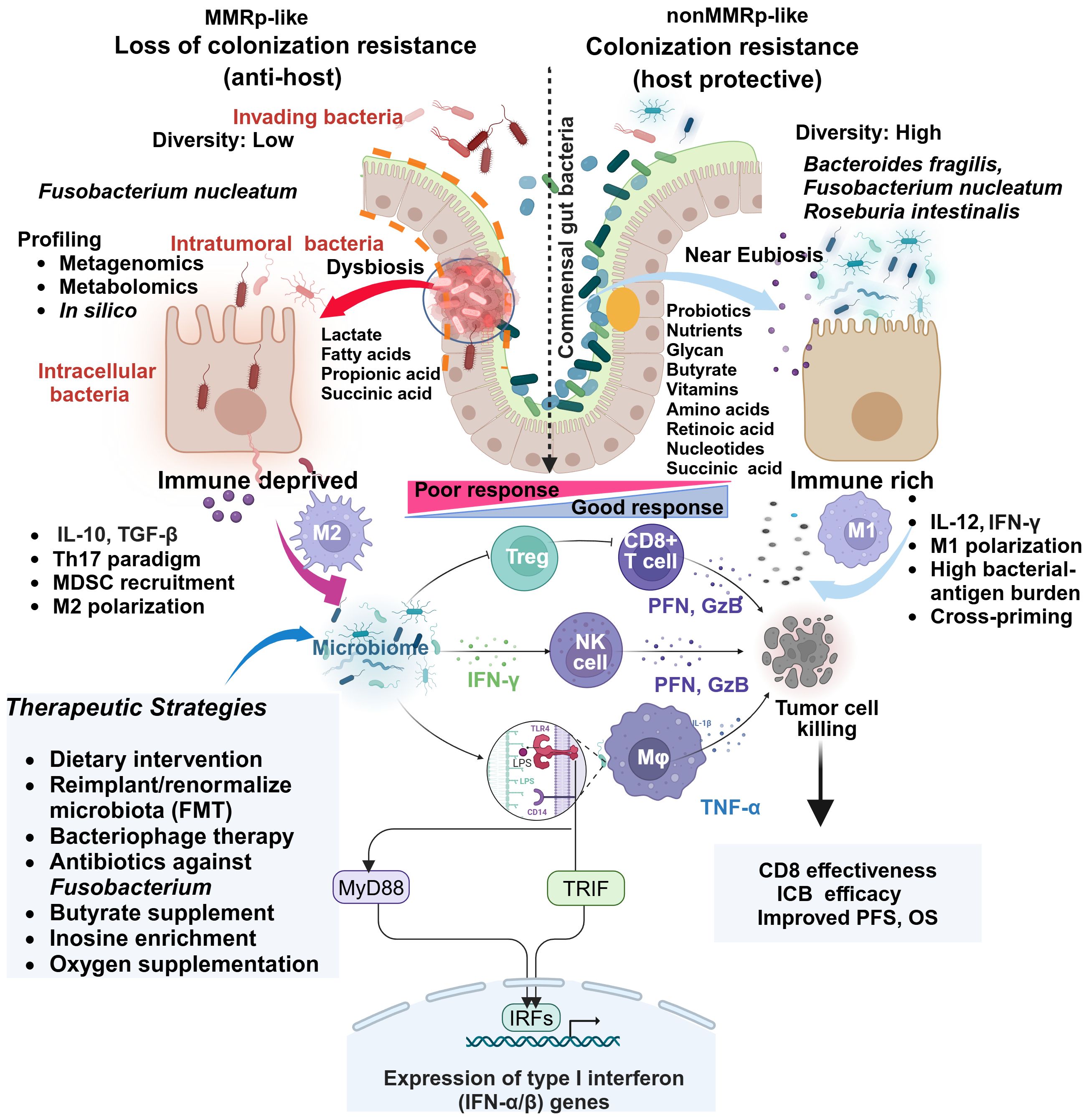
Figure 2 Comparative gut microbiome profiles in MMRp and non MMRp tumor hosts underline multiple contextual constraints and explain barriers to therapy success. The global loss of protective gut resident commensal bacteria makes the border porous for invading bacteria and supports their colonization. Different next-generation sequencing platforms and in silico analysis enable determining the high load of such bacteria and dissecting the loss of diversity in MMRp interface. This maladaptation promotes the metabolic bias in the microenvironment characterized by the overproduction of lactate, propionate, long-chain fatty acids and concomitant loss of glycans, short-chain fatty acids like butyrates, vitamins, amino acids, retinoic acids, and nucleic acids. In such conditions, intra-tumoral and intracellular bacteria facilitate the polarization of immune cells like MDSC, Treg and M2 macrophages, creating a suppressive paracrine cytokine loop. This polarization indicates a sharp contrast with MMRd, where a permissive metabolic footprint favors the preservation of cytokines like IL-12 and IFN-γ, bacterial antigen presentation by M1 macrophages. Under this condition, the interaction of bacterial LPS with TLR in macrophages triggers a signaling pathway via canonical myeloid differentiation primary response 88 (MyD88) and TIR domain-containing adaptor inducing interferon-β (TRIF) that engages IRFs and produces type 1 IFN. Fn-mediated altered cytokines and other anti-apoptotic mechanisms confer resistance by orchestrating an M1 toM2 paradigm shift. Macrophages (M1), CD8, and NK-mediated production of anti-tumor cytotoxic effectors like perforin (PFN) and granzyme-B (GzB) elicit tumor-killing effects. TGF-β, IL-10 and IL-17 impair immune-effector function in MMRp. Multiple strategies focusing on improving the hostile tumor-microbiome interface in MMRp can reverse the suppressive state. Adapted from “Keystone Gut Microbiota Species Provide Colonization Resistance to Invading Bacteria” by BioRender.com (2021). Retrieved from, https://app.biorender.com/biorender-templates.
In a permissive MMRd ecosystem, microbiota supports vitamin A metabolite and retinoic acid accumulation. It galvanizes nucleic acid and protein breakdown machinery in a heterogenous gut immune-interaction network. The accumulation of lactate and other short-chain organic acids like propanoic acid, owing to microbiota depletion in MMRp, makes the tumors immunosuppressive (109) (Figure 2). Lactic acid-producing bacteria Lactobacillus iners rewire host tumor metabolic pathway in cervical cancer and confer chemo- and radiotherapy resistance. A similar L-Lactate producing bacterial population reduces recurrence-free survival (RFS) in colorectal adenocarcinoma (110). Targeting the metabolic hardwire that regulates local oxygen levels or reduces hypoxia will provide insights into their therapeutic prospects (111, 112). For example, antagonists targeting immune-specific CD73 or genetic deletion of A2AR, reversing lactate and hypoxia-induced immune suppression are in clinical development (113, 114). Therefore, combining such agents with approved immune or non-immune therapies may boost the anti-tumor response in MMRp. A spatial metabolomics landscape of the tumors also adds an interactive milieu in this context (115). Extending its crosstalk with the microbial metabolomic network will further define the metabolic vulnerabilities in MMRp and other similar tumors.
4.4 Ecosystem deep mining in MMRp precision microbiomeWhile the proximal and distal gut microbiota define the fate of tumors, intra-tumoral bacteria in such scenarios pose a serious health challenge. Integrated metagenomics and metabolomic profiling further expand the scope of ecosystem-level deep mining and shed light on undetected metabolites (116, 117). Pan cancer profiling of intra-tumoral bacterial hubs helped elucidate their distinct indication-specific composition. It also confirmed their intracellular presence, which covers tumor and immune cells. For example, Firmicutes and Bacteroidetes phyla were the two most abundant species in a cohort of 22 CRC samples (118). From these perspectives, the distinct orientation of dysbiosis and its polymorphic microbiome spectrum are suspected to drive toxin-induced mutagenesis in the gut ecosystem (119). CRC patient-derived fecal gavage has been associated with inducing GI tract carcinogenesis in germ-free mice (120). Contrary to this, CRC with MMRp persuasively displayed a hostile metabolic microenvironment that facilitates disease progression and therapy resistance. The conceptual progress in tumor-microbiome interactions also sheds new light on the presence of intertumoral bacteria and their geospatial micro-niche in the tumor ecosystem. New single-cell RNA sequencing technology has mitigated the low biomass challenge and improved the robustness of capturing the tumor microbiome diversity. The preferentially high bacterial population density in vasculature deprived (i.e. CD34 negative) and Ki67 negative pockets with suppressive immune contexture in CRC forms the basis for the non-random heterogeneity of microbiota (121). This profiling added valuable knowledge about resources in such tumor ecosystems and can gauge potential benefits from complementary therapy.
Intra-tumor microbiome maps of CRC (and GI) have been developed in a pan-cancer study. It revealed an MSI-MSS distinction of their communities. It also showed poor survival after ICB in the F. nucleatum high group in the case of NSCLC (109, 122). However, there is a paucity of knowledge on the premises of MMRp (102, 118). As a dysregulated immune-microbiome interplay in MMRp CRC corrupts TME, reinstating a patient-friendly microbiota context and relevant therapeutic strategies potentiate a better therapy response in MMRp (109, 122, summarized in Figure 2 and Table 2). Further studies on the intracellular bacterial population in MMRp+ CRC and its spatial distribution in an immune-excluded context coupled with metabolic programming will offer valuable insights and perspectives. The comprehensive landscape of tumor-associated bacteria highlights the scope of fine-tuning therapeutic intervention at the tumor-microbiome interface.
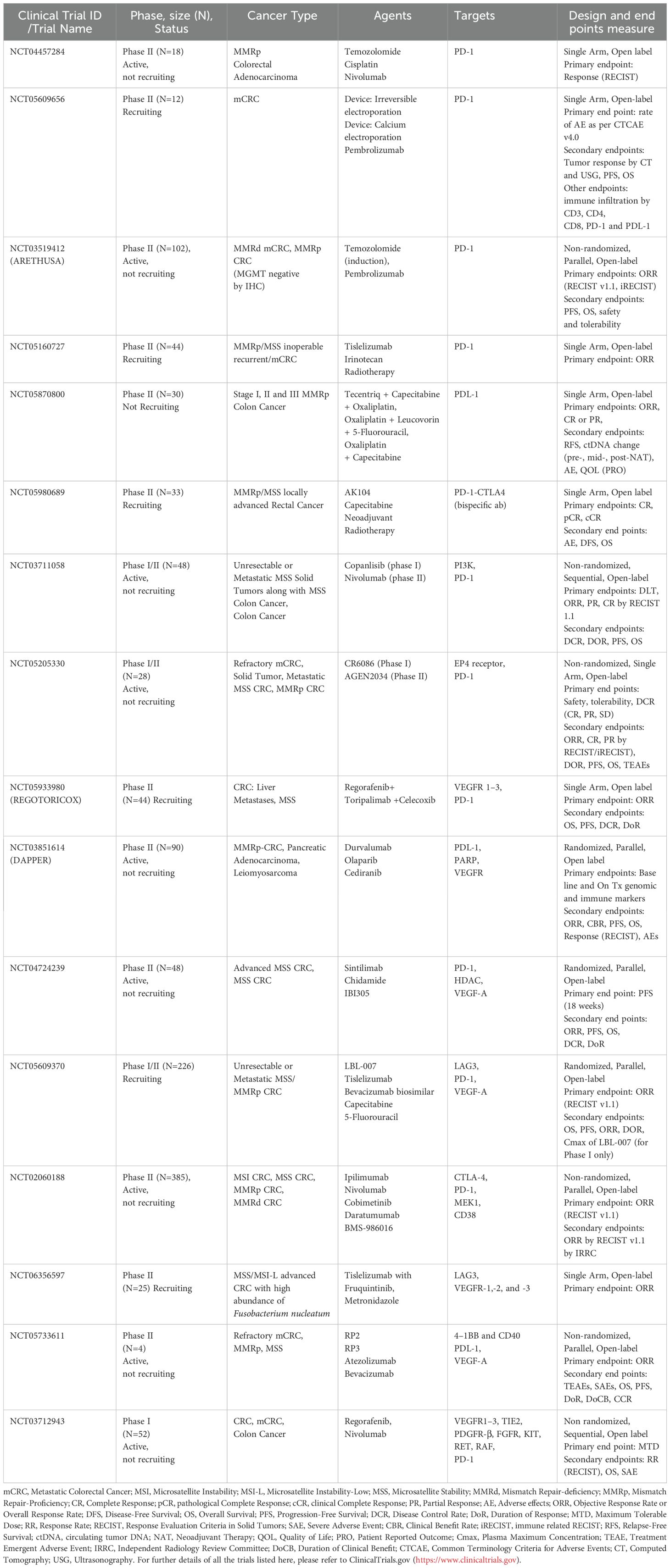
Table 2 Ongoing clinical trials that include MMRp positive CRC patients.
4.5 Therapeutic strategies in reversing onco-microbiome niche in MMRpSeveral strategies leveraged diverse aspects of microbiomes and their perturbation in preclinical and clinical settings and identified their potentials and limitations.
4.5.1 Probiotic gut bacteria in metabolic immunomodulation in MMRpThe probiotic bacterium Clostridium butyricum inhibits Wnt signaling, reduces the risk of colon cancer development and boosts anti-microbial macrophage function while sparing inflammation-induced tissue damage (123, 124). Faecal microbiota analysis of CRC patients revealed that Roseburia intestinalis, a probiotic species, and the metabolite (butyrate) generated by it, protect mice (CT-26 and MC 38) and human hosts from gut inflammation and damage. They also unleash CD8-induced anti-PD-1 efficacy in MSI-low/MMRp (CT-26) orthotopic mice. The substantial depletion of this species was observed in patient-derived stools compared to healthy individuals. Its transfer to mice from healthy humans inhibited tumor growth (125). A conversation between Group 3 innate lymphoid cells (ILC3s) and T cells through the engagement of MHCII complex following supplementation of microbiota orchestrate type 1 innate immune response and responsiveness to PD1 inhibition in mice. Lack of MHC-II in ILC3s or microbiota harvested from ILC3s dysregulated subjects failed to elicit immune therapy response following their transfer to mice (126).
4.5.2 Microbiome as an adjuvant in immune checkpoint therapySimilar systemic clearance was reported when Fn was targeted by silver nanoparticle-bound M13 phage (Ag-M13). Reduction of Fn-induced MDSC and reinvigoration of APC functions were reclaimed under this therapy condition in mice models. Ag-M13 acted in synergy with ICB or chemo-agents (127). In general, antibiotics also have precedence in interfering with ICB (128). The dose, sequence, spectrum, limited and emergency-only use of antibiotics can overcome this challenge. Target-specific antibiotics and bacteriophages make their way to better deal with this situation (66, 129). Through modulating specific chemokine production, gut microbiota helps infiltrate the anti-tumor T cells to the tumor sites and improves the survival opportunities (130). Bifidogenic live bacterial products complementing the microbiome, tested initially in renal cell carcinoma (RCC), would add an interesting vertical in this direction (131). More importantly, several vaccines targeting Fn and Bf in colon cancer are under preclinical development. Depending on the risk association, these vaccine candidates will be used for therapeutic or prophylactic purposes (132).
4.5.3 Microbiome guided therapyAnalysis of drug-metabolome association in allogeneic hematopoietic cell transplant (alloHCT) recipients among cancer patients may benefit these populations. Longitudinally tracked fecal microbial species revealed a substantial loss of alpha diversity or dysbiosis. It also gathered information regarding a reversal under different medications. An in silico computational prediction model mirrors the in vitro measurement of antibacterial activity and patient clinical outcomes (133).
Antibiotics can reduce bacterial loads that pose a threat to prognosis and response to therapy. For example, F. nucleatum in CRC accumulates succinic acids. This high succinic acid in tumor hinders response to PD1 inhibition by obstructing CD8 cells. Both FMT from the responder and antibiotic metronidazole overcome this restrain (66). Likewise, by enhancing the safe and effective local delivery, liposomal antibiotic administration in mice targeting F. nucleatum elicited cytotoxic T cell response through increasing the immunogenic neoantigen burden of bacterial origin. This modulation further helps in T cell priming and recognition of antigen-naive and reactive tumors (134) (Figure 2). In clinical CRC, before surgical resection, eliminating anaerobic bacteria load upon antibiotics treatment improved disease-free survival (DFS) by 25.5%.
Antibiotics can lead to the vertical loss of healthy intestinal flora, but probiotic and fecal microbial transplantation (FMT) can compensate for the antibiotic-induced loss of gut microbiota. Current limitations within this realm involve compatibility, stability, unknown composition, kinetics, and dynamics, which could be ethical concerns. For an amenable resolution of these concerns, instead of adopting a blanket use, individualized assessment of gut health and other supporting methods like physical activities are important factors that can improve the outcomes (135–137). Reimposing anti-dysbiotic barriers requires coordinated approaches. Supplementing the gut ecosystem with niche-modifying commensal species prevents colonization by invaders. It prevents the accumulation of metabolically challenging pathogenic microbes and releases bacterial antigens to boost the pro-immunogenic immune network (137) and Figure 2.
5 Response prediction biomarkers for improving therapy outcomes in MMRpBoth immune and targeted therapy rely on the individualized selection of patients to maximize benefit from a given treatment modality. Integrating biomarkers that predict response is of pivotal importance in this context. This also provide information pertaining to resistance and help designing rational therapeutics.
5.1 Immune response in MMRp and mechanistic underpinningsCollectively, 85% of CRCs are of MMRp type (138). Overall, 10% CRC and 5% metastatic CRC of MMRp status show a response to ICB (33). Combining anti-PD1 with novel anti-CTLA4 in heavily pretreated (median prior line: 4) CRC with MSS status showed promising safety and efficacy (ORR 24%). In case of no history of liver metastases/ablation of liver metastases without recurrence, a better outcome was achieved (n=24, ORR 42%, and DCR 96%). This response included a patient with SD (RECIST 1.1) who showed ongoing metabolic complete response (mCR) by PET after CEA normalization. For all responder cases, metastatic sites spanned soft tissue, peritoneum, retroperitoneum, pleural effusions, bone, lungs, and lymph nodes. Responder mutation profile confirmed RAS mutations (4 KRAS, 1 NRAS), no BRAF mutations, a high TMB (TMB=10) in one case, one case of CPS >50%, and no single POLE mutations cases (139). Both MSS CRC with and without liver metastasis showed benefits from ICB, where liver metastasis conferred more frequent resistance (140). This implies the urgent requirement to improve the overall response to this therapy in MMRp tumors, reducing its gap with MMRd. Indeed, challenging the current response rate for all modalities with new and more effective treatment regimens is a continuous process and needs innovative, rational approaches integrated with multimodal diagnostics and predictive tools.
5.1.1 IL-17 and LAG-3 are therapy barriersFrom oncoimmune perspectives, however, response predictive gene signatures revealed that a preexisting immunoreactive profile does not explicitly depend on suppressive tumor immune microenvironment represented by spatial CD8 and IFN-γ and colocalized PDL-1/IDO1 checkpoint genes. Irrespective of IL-17 low or high niche states, the IL-17 low MMRp landscape mimicked a primary CRC responsive to ICB. In the same study, a panel of immunomodulatory genes (precisely, LAG-3, CD8A, CD4, CD274) showed similar expression patterns between MMRp and MMRd responder cohorts. However, it indicated reciprocal downregulation in the MMRp non-responder cohort (67). Rationally targeting the IL-23/Stat3/IL-17 signaling axis in IL-17 high MRRp+ CRC may offer a mechanistic basis for overcoming adaptive resistance to ICB. Analysis of TCGA data and cell line profiling of MMRp from CRC revealed that high expression of immunoglobulin superfamily 6 (IGSF6) is correlative with infiltration of CD4+ T cells, CD8+ T cells, CD68+ macrophages and conferred sensitivity to immunotherapy and chemotherapy (
留言 (0)