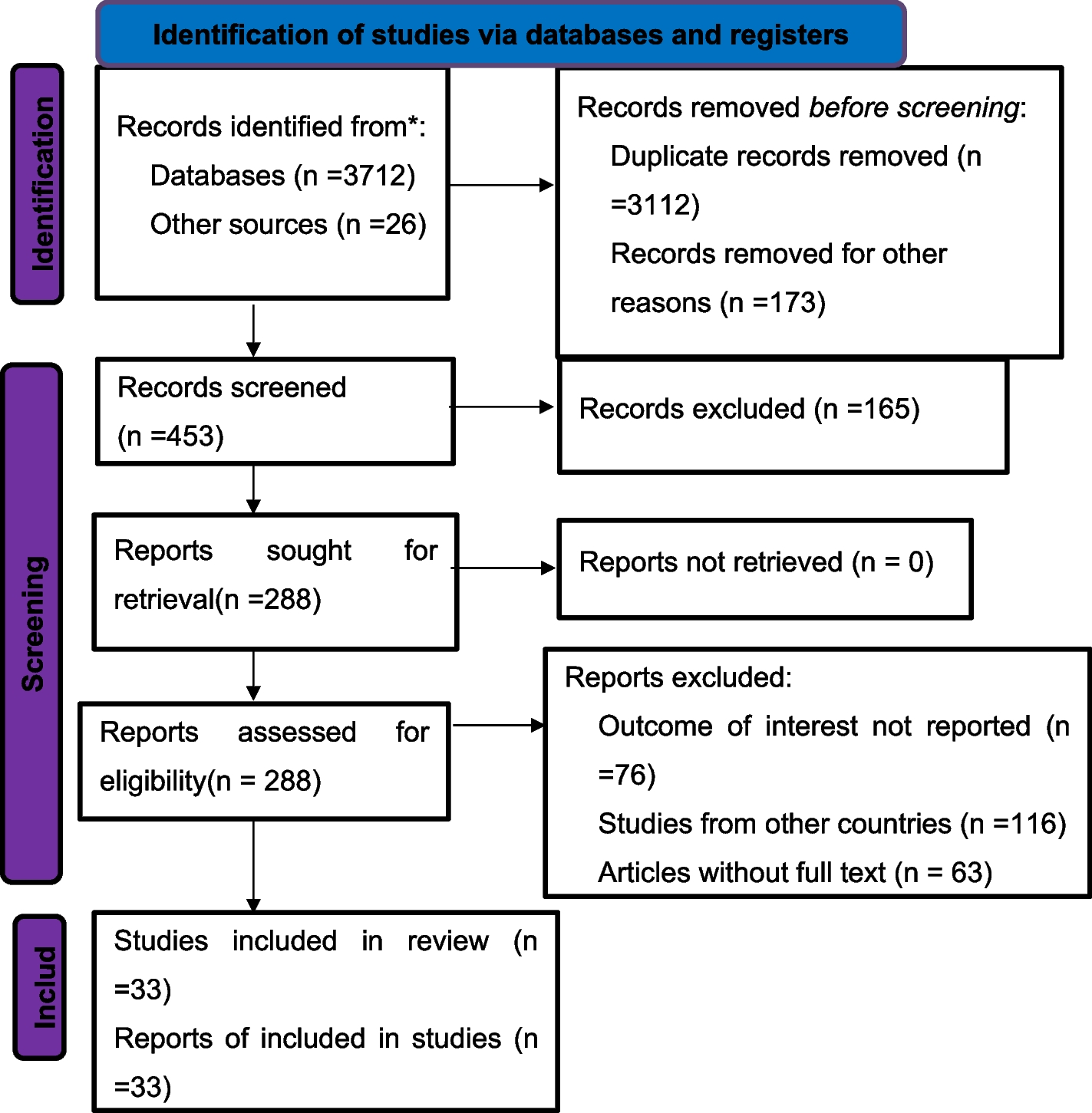
This study reported two patients with X-ALD, whose initial symptoms were adrenal insufficiency and were clinically diagnosed with Addison’s disease (primary adrenal insufficiency). However, the possibility of X-ALD (Addison’s-only) was ignored, and the disease was not monitored in time. Neurological symptoms after infection led to further brain MRI and genetic testing, and the two patients were finally diagnosed with X-ALD (CCALD). As demyelinating lesions may not appear rapidly within days of infection, the patients may have progressed to CCALD prior to infection, and the infection may have exacerbated the inflammatory demyelination of CCALD and triggered the emergence of neurological symptoms. This study highlights the importance of genetic testing in patients with adrenal insufficiency and the role of infection in promoting cerebral ALD.
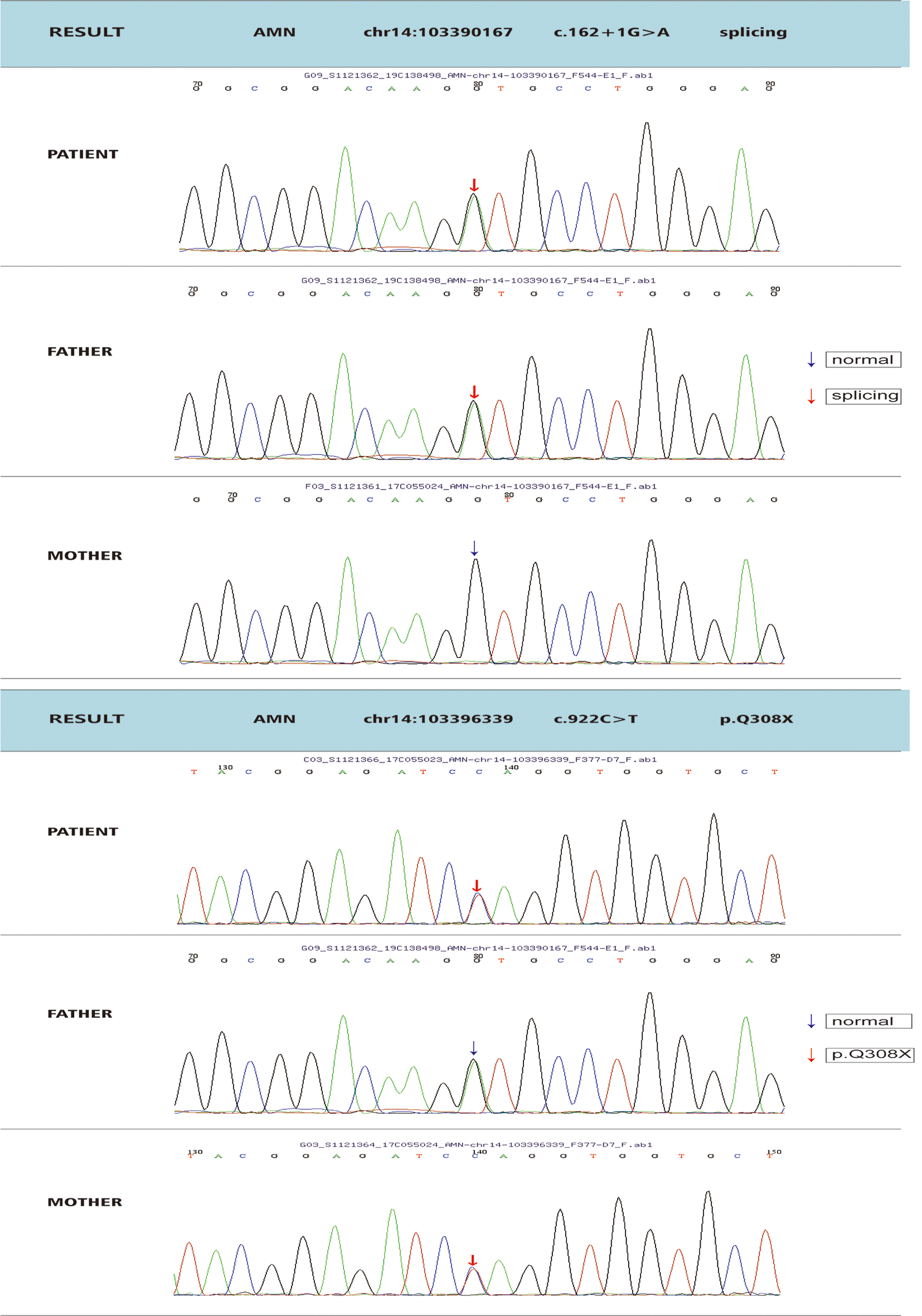
X-ALD is peroxisome disease caused by ABCD1 variation. Its clinical phenotypic severity varies greatly, with mild cases presenting only adrenal insufficiency, that is, Addison’s-only, and severe cases presenting rapidly progressive CCALD [3, 7]. Adrenal insufficiency can be the first clinical manifestation of X-ALD. For CCALD, it may only present as adrenal insufficiency before neurological symptoms [4, 23,24,25]. During this period, given that only adrenal insufficiency symptoms were present, such as skin and mucosal pigmentation, but no neurological symptoms, it was easy to be simply diagnosed as Addison’s disease (primary adrenal cortical insufficiency). Meanwhile, the possibility of ABCD1 variation-related X-ALD (Addison’s-only) was ignored, especially the risk of progression to cerebral ALD [1, 26,27,28]. In this study, two patients initially had only skin and mucosal pigmentation. They were clinically diagnosed with Addison’s disease and only received symptomatic treatment with hydrocortisone. Given its incomplete genetic testing, the existence of X-ALD (Addison’s-only) was ignored. Therefore, skin and mucosal pigmentation is also an early warning symptom of X-ALD. Thus, the possibility of X-ALD must be considered, and genetic testing must be completed as soon as possible.
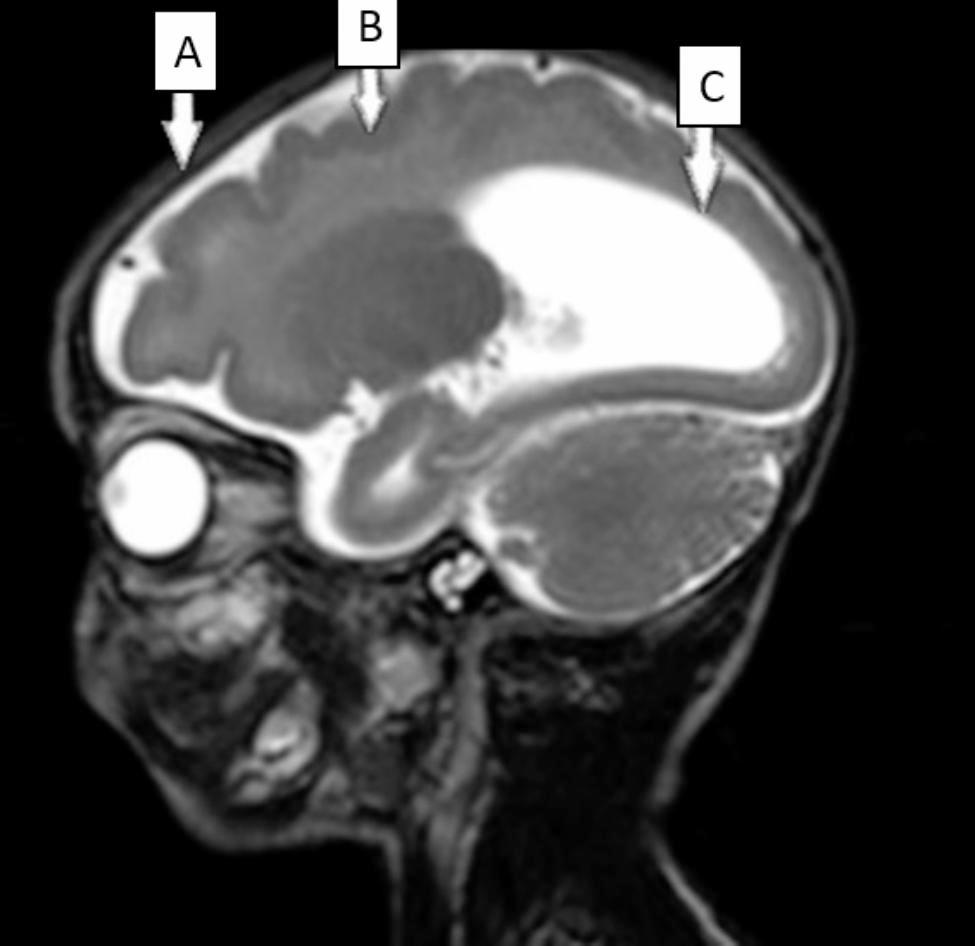
For CCALD, before the onset of clinical symptoms, the earliest brain abnormalities are demyelinating lesions confined to brain MRI, which means the patient is in a presymptomatic stable state. Neurologic symptoms may occur when inflammatory demyelination progress. Once neurological symptoms appear, the disease progresses rapidly, and the presymptomatic stable state is difficult to maintain. Affected patients may have difficulty in communication, spasmodic gait. Eventually, patients may develop major functional dysfunction (MFD), such as being bedridden, blind, unable to speak or respond, fed by gastrostomy; moreover, death usually occurs two to four years after onset of symptoms [29]. Therefore, delaying the onset of neurological symptoms is critical to stabilizing the condition. In this study, after the infection of influenza A virus, the ACTH level of patient 1, which had dropped to normal, increased again and gradually manifested neurological symptoms of CCALD. Patient 2 also presented with neurological symptoms after respiratory virus infection. Consistently, both patients presented an onset of neurological symptoms within days of infection. This fulminant clinical process is mainly associated with the aggravation of inflammatory demyelinating lesions in the white matter [18, 19]. The two patients underwent brain MRI after neurological symptoms, and the scan indicated demyelinating lesions. As demyelinating lesions of the brain may not appear rapidly within a few days after infection, the demyelinating lesions on brain MRI may have existed before infection. Both patients had progressed to CCALD prior to infection and were in a pre-symptomatic stable state (only demyelinating lesions in head MRI). The infection event aggravated the demyelinating lesions and accelerated the appearance of neurological symptoms. Considering the role of environmental factors in the pathogenesis of X-ALD [18, 19], the experiences of these two patients suggest that infection, as a triggering factor, may increase the risk of triggering progression of inflammatory demyelinating in patients with CCALD.
We further analyzed the possible pathological basis of CCALD triggered by infectious factors. VLCFA-associated oxidative stress in the brain leads to inflammatory demyelination in patients with ABCD1 variations [8, 9]. Similarly, after infection, given increased body metabolism, hyperactive mitochondrial metabolism produces more ROS, which may increase white matter susceptibility to oxidative stress [8]. In addition, infections, such as viral infections, can activate the immune response and promote the release of inflammatory factors [30]. So, infection-induced oxidative stress and inflammation may be important factors in accelerating the progression of inflammatory demyelination. In addition, considering the higher vulnerability of brain microvascular system to ABCD1 deficiency in the setting of inflammation [20,21,22], the inflammatory response caused by viral infection may further increase BBB permeability and microvascular flow heterogeneity, thus leading to the infiltration of peripheral inflammatory cells and brain parenchymal damage. Therefore, for patients with CCALD, infection, as a hit factor, promotes the progression of inflammatory demyelination of the brain. For CCALD, after early diagnosis, patients should be protected as extensively as possible to avoid infection, head trauma, and other events, which can stabilize the disease to a certain extent, to reduce or delay its progression to MFD or death.
Hematopoietic stem cell transplantation (HSCT) or hematopoietic stem cell gene therapy can effectively delay the progression of neurological diseases only in the early stage of CCALD [16, 31]. More importantly, better clinical outcomes can be achieved by receiving HSCT before the onset of neurological symptoms [11]. On this basis, neonatal ALD screening has become popular in recent years [32, 33]. Aubourg et al. recommend that any boy or adult male with Addison’s disease must be tested for ALD, given the genetic counseling, and the potential benefits of therapeutic intervention [34]. Given the prognostic implications of undiagnosed ALD, genetic testing should be performed as soon as possible for patients with early adrenal insufficiency related symptoms to achieve early diagnosis of ALD and improve the general outcome of these patients. If a definite diagnosis of X-ALD is made, brain MRI should be monitored regularly. Once inflammatory demyelinating lesions associated with cerebral ALD occur, treatment should be given as soon as possible. In addition to early treatment, this study also emphasizes early prevention before the onset of neurological symptoms, that is, avoiding the occurrence of hit events, such as infection, to delay the progression of the disease.








留言 (0)