
記住我
In the 1940s, first anthracycline, Daunorubicin (DAU), was discovered by the Streptomyces bacteria, marking a significant breakthrough in oncology (Marco et al., 1981). Clinical trials demonstrated remarkable success in using DAU to treat acute leukemia (Tan et al., 1967). Subsequently, the exploration of other anthracycline antibiotics led to the discovery of the anticancer drug doxorubicin (DOX), a precursor of DAU, which not only proved effective for leukemia but also exhibited therapeutic properties against most solid tumors (Arcamone et al., 1969). Currently, widely used anthracycline compounds in clinical practice include DOX, Pirarubicin (THP), Idarubicin (IDU), and Epirubicin (EPI). Anthracyclines remain frontline agents in many cancer chemotherapy regimens.
However, despite their ability to improve long-term survival in cancer patients, anthracycline-type drugs can induce cardiotoxicity after reaching a certain cumulative dose (Sawicki et al., 2021). Middleman observed a dose-dependent relationship between anthracycline drug dosage and cardiac damage in 1971 (Middleman et al., 1971). Clinically, acute anthracycline-induced cardiotoxicity is characterized by electrocardiographic abnormalities (20%–30%), neutropenia (6%), arrhythmias (3%), and in some patients reversible cardiac insufficiency (Middleman et al., 1971). Anthracycline-induced chronic cardiotoxicity may occur months or years after initial therapy and can be categorized as early-onset or late-onset. Early-onset chronic cardiotoxicity usually occurs within 1 year after completion of anthracycline therapy in adults, whereas late-onset chronic cardiotoxicity may occur decades after treatment and is commonly seen in pediatric patients who have received chemotherapy (van Dalen et al., 2011). The incidence of doxorubicin-induced cardiotoxicity, primarily congestive heart failure, is strongly dependent on the cumulative dose. Studies have shown that the incidence of congestive heart failure was 3%, 7%, and 18% at cumulative doses of 400, 550 and 700 mg/m2, respectively. A combined analysis of three studies of 630 patients with breast and lung cancer further showed that the incidence of congestive heart failure was even higher, with rates of 4.7%, 26% and 48% at cumulative doses of 400, 550, and 700 mg/m2, respectively (Silber, 2004). Due to the limited terminal differentiation of cardiac myocytes, they are vulnerable to the effects of anthracycline drugs, leading to cardiac injury (Khouri et al., 2012).
2 Types of cardiac damage induced by anthracycline-type drugsCardiac damage caused by anthracycline-type drugs is the result of various molecular processes, primarily centered on alterations in cardiac myocyte function and pathological cell death (Nebigil and Désaubry, 2018). The initial manifestation of cardiac damage is subclinical myocardial injury, progressing to early asymptomatic reductions in left ventricular ejection (LVEF) fraction and ultimately leading to severe heart failure (Yeh and Bickford, 2009) (Figure 1).
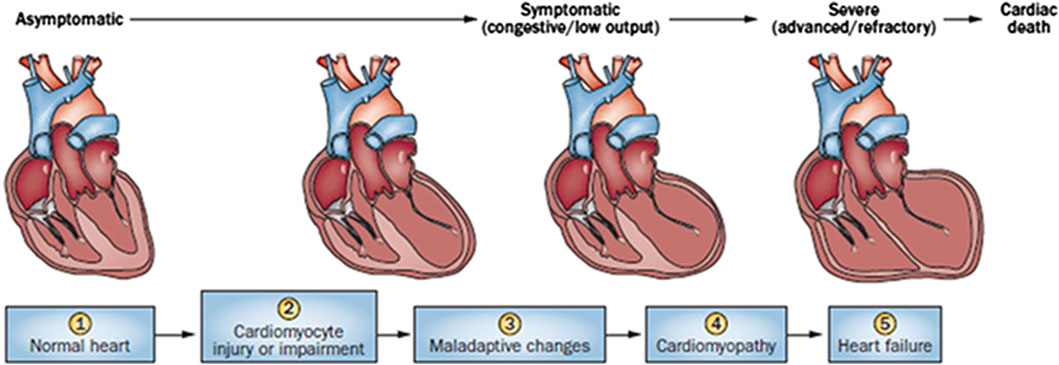
Figure 1. Stages in the course of Anthracycline-induced ventricular dysfunction. (1) Primary prevention is possible at this stage by reducing risk factors in high-risk populations (such as those receiving anticancer therapy). (2) Secondary prevention is possible at this stage to reduce the effects of the treatment-induced injury. (3) Secondary prevention is also possible at this stage. (4) Clinically significant conduction and rhythm abnormalities might be observed. (5) Radical therapies might be required at this stage (such as heart transplant) if there is failure of medical management. Preventive strategies are progressively less effective as the toxicity increases. Treatment strategies have a greater impact when used to treat themore-diseased heart, but have longer effects if initiated early.
Anthracycline-induced cardiac damage can be categorized as acute and chronic, depending on the initial symptom presentation (Cardinale et al., 2015). Acute cardiac injury typically manifests within the first few days of administering anthracycline drugs, presenting as acute myocarditis with myocardial cell injury, inflammatory infiltrates, and interstitial edema (Ferrans, 1978). Clinically, acute cardiac damage manifests as changes in electrocardiograms, neutropenia, arrhythmias, and, in some patients, reversible cardiac dysfunction (Shan et al., 1996). Chronic cardiac damage can develop months or years after initial treatment and can be classified as either early or late onset. Early-onset chronic cardiac damage usually arises within the first year following anthracycline drug treatment, whereas late-onset chronic cardiac damage can manifest decades later, particularly in pediatric patients. (Cardinale et al., 2015). Chronic cardiac damage is characterized by cardiomyopathy or congestive heart failure (CHF), along with cardiac enlargement, reduced LVEF fraction, ST-T segment changes, and cellular-level features such as cytoplasmic and mitochondrial swelling, organelle rupture, cardiomyocyte death, and myofibrillar disarray leading to cytoplasmic vacuolization (Berry and Jorden, 2005). When the cumulative dose of anthracycline drugs reaches 550 mg/m2, approximately 26% of patients experience irreversible LVEF reduction, and more than 7% develop heart failure (Swain et al., 2003).
3 Molecular mechanisms of anthracycline-induced cardiac damageCurrently, the primary mechanism of anthracycline-induced cardiotoxicity is believed to involve the inhibition of Topoisomerase (TOP) 2β (Henriksen, 2018). Other mechanisms include disruptions in iron ion metabolism, mitochondrial dysfunction, myocardial fiber dissolution, sarcoplasmic reticulum calcium imbalance, oxidative stress, and cell apoptosis (Bansal et al., 2019). While many mechanisms have been associated with cardiac toxicity, the exact mechanisms remain unclear.
3.1 TOP inhibitionAnthracycline drugs act by binding to DNA TOPs, inserting into DNA, promoting DNA strand breaks, and causing cardiac damage (Tewey et al., 1984). TOPs are enzymes that regulate DNA replication, transcription, recombination, and chromatin remodeling by inducing temporary single or double-strand breaks when they open DNA strands. In human cells, there are two Top2 isoforms, Top2α and Top 2β. Top2α is expressed at high levels during the cell cycle in undifferentiated and proliferating cells, including those of tumors, with varying levels. In contrast, Top2β is expressed in quiescent and differentiated cells, such as those found in cardiac muscle, maintaining consistent levels throughout the cell cycle (Wang, 2002) (Figure 2).
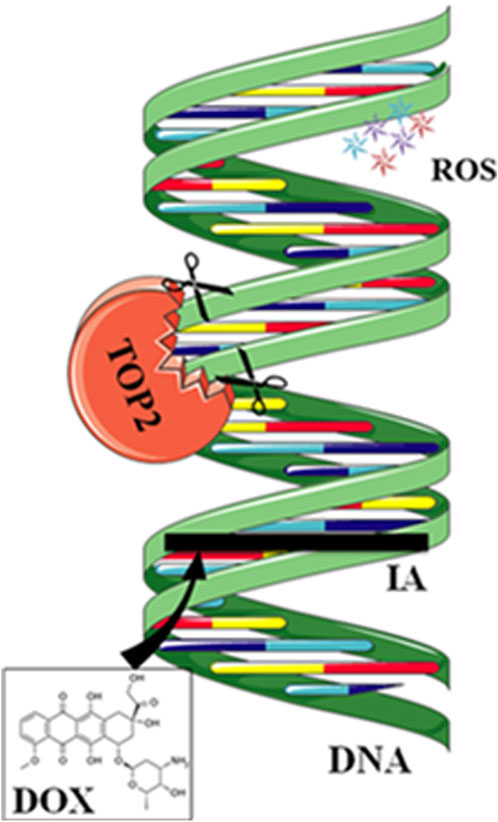
Figure 2. Mechanisms of action of doxorubicin (DOX). DOX intercalates between strands of DNA double helix. The formation of a ternary complex (TOP2-DOX-DNA) prevents enzyme turnover. The latter blocks the catalytic cycle after DNA is cleaved and before DNA relegation. DOX biotransformation results in the formation of ROS.
DOX binds to Top2α, arresting tumor cells in the G1/G2 phase and inhibiting DNA replication, leading to apoptosis. However, anthracycline drugs also bind to Top2β, inhibiting DNA replication, resulting in apoptosis of cardiac muscle cells. Experiments have shown that specific knockout of the Top2β gene in mouse hearts can alleviate DOX-induced heart failure. Compared to the control group, double-strand DNA breaks decrease, suppressing apoptosis (Zhang et al., 2012). DZR besides its iron-chelating function, can inhibit Top2β activity, which may contribute to its cardiac protection (Vavrova et al., 2013). Crystal structure analysis shows that DZR stabilizes ATP in the ATPase domain of Top2β, preventing the reopening of the Top2β subunit and locking the Top2β monomer in a closed dimeric structure. This, in turn, prevents anthracycline drugs from binding to the TOP-DNA complex and inducing DNA damage. (Classen et al., 2003; Lee et al., 2017). Researchers have developed anthracycline compounds that specifically target Top2α. While this approach does not affect the expression of Top2β in differentiated cardiac muscle cells, it may potentially impact undifferentiated cardiac progenitor cells expressing Top2α.
In addition to expressing Top2β, cardiac muscle cells also express mitochondrial TOP 1 (Top1mt), which regulates the stability of mtDNA. Mice that lack Top1mt demonstrate mitochondrial defects, such as compromised synthesis of respiratory proteins and disrupted mitochondrial ultrastructure. They are more susceptible to DOX-induced damage compared to normal mice, leading to cardiac injury (Khiati et al., 2014). Therefore, Top1mt has a cardioprotective role, and genetic variations in Top1mt may increase sensitivity to anthracycline cardiotoxicity.
3.2 Iron ion metabolism disturbanceUnder normal physiological conditions, there is only a minimal amount of biologically active free iron in cardiac muscle cells. Most iron ions are bound to iron proteins to prevent their escape and avoid tissue and cell damage. When there is an excess of iron ions in the cells, anthracycline drugs increase intracellular reactive oxygen species (ROS) and induce oxidative stress reactions, causing cardiac muscle damage (Xu et al., 2005).
When a significant amount of anthracycline drugs accumulates within cells, the anthracycline drug’s core structure, anthraquinone, can be reduced to semiquinone radicals by reduced nicotinamide adenine dinucleotide phosphate dehydrogenase (NADPH). Nucleotide phosphate dehydrogenase (NADPH) to semiquinone radicals. These radicals undergo auto-oxidation under aerobic conditions, generating anthraquinone radicals and superoxide anions (O2-), leading to intracellular O2- accumulation. Meanwhile, O2- is converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). H2O2 can react with iron ions through the Fenton reaction, converting H2O2 into toxic hydroxyl radicals (-OH), thus causing sustained increases in intracellular ROS levels and oxidative stress reactions, leading to cardiac muscle damage (Doroshow and Davies, 1986; Winterbourn, 1995).
Anthracycline drugs can also directly interact with iron ions, forming complexes in a cyclic manner between Fe2+ and Fe3+, leading to oxidative stress and mitochondrial dysfunction (Myers et al., 1982). DOX derivatives can also chelate iron ions from cytoplasmic iron-regulatory protein 1 (IRP1) iron-sulfur (Fe-S) clusters, rendering IRP1 inactive. Inactive IRP1 increases the synthesis of transferrin receptor (TF), reducing the synthesis of intracellular iron storage protein (ferritin), disrupting cellular iron homeostasis, ultimately resulting in continued increases in intracellular free iron (Minotti et al., 1998) (Figure 3).
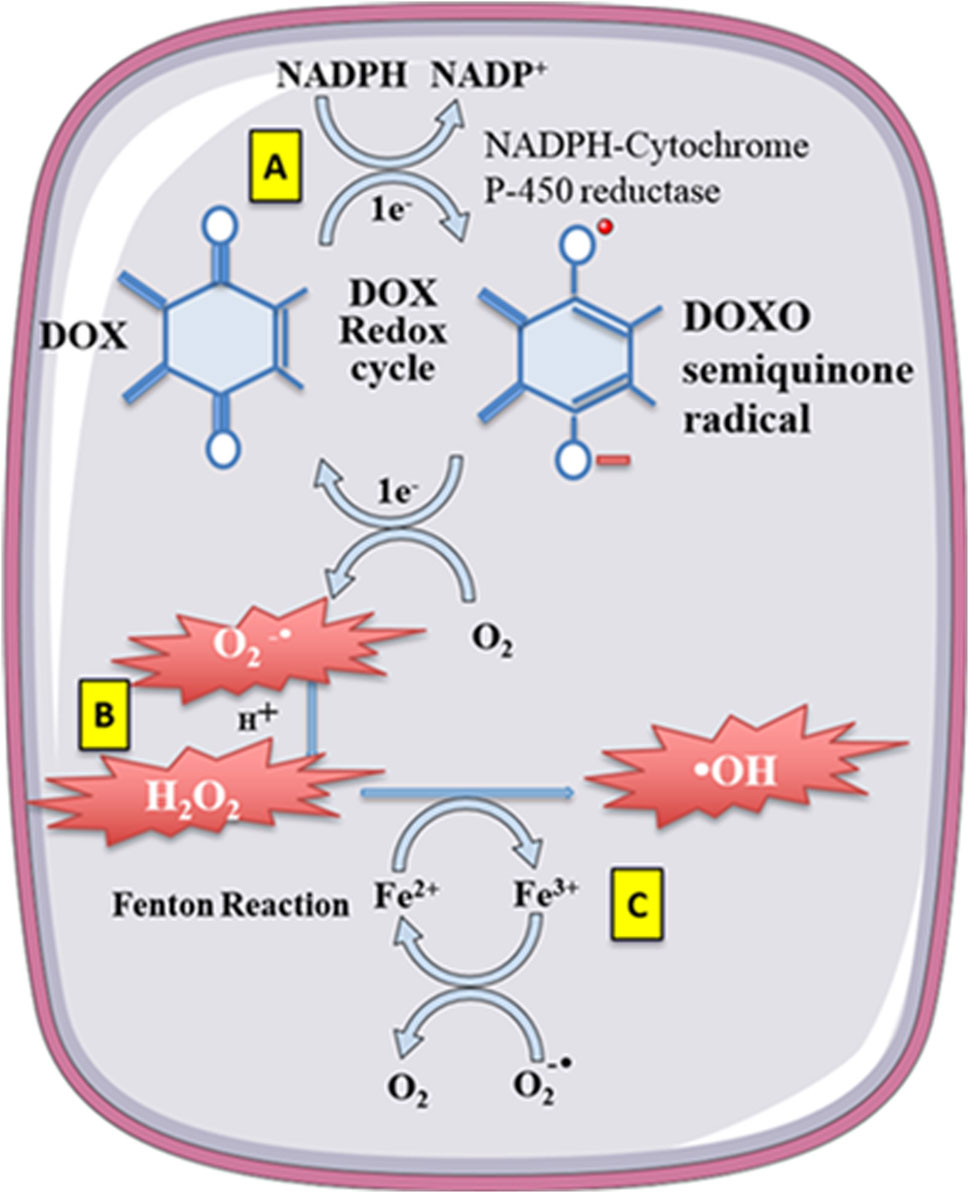
Figure 3. Doxorubicin (DOX) undergoes redoxcycling catalysed by NADPH-Cytochrome P-450 reductase. (A) The One-electron (1e-) reduction of the quinone compound leads to redox cycling and generation of superoxide anion radical. (B) O2-undergoes dismutation to form H2O2 either spontaneously or catalysed by superoxide dismutase. (C) H2O2 then reacts with the transition metal ion Fe2+, giving rise to -OH. Free Fe2+ is toxic to cells as catalyst in the formation of free radicals from ROS via the Fenton reaction.
Mitochondrial iron export protein (ATP Binding Cassette Transporter B8, ABCB8) is one of the essential factors affecting mitochondrial iron metabolism during DOX treatment. Studies have reported that DOX can downregulate the expression of ABCB8, leading to increased iron accumulation within mitochondria. Transgenic mice overexpressing ABCB8 exhibit protective effects against DOX-induced cardiac muscle damage. ABCB8 overexpression reduces the iron content in mitochondria, thereby reducing DOX-induced cardiac toxicity (Menon and Kim, 2022). This suggests that reducing intracellular mitochondrial iron ion content may be an effective strategy to prevent anthracycline-induced cardiac toxicity.
Although both in vitro and in vivo studies confirm an increase in free iron content within cardiac muscle cells after anthracycline treatment, the role of iron disturbance in inducing cardiac damage is still debated. Not all iron chelators have shown efficacy in mitigating anthracycline-induced cardiac damage (Stěrba et al., 2013). DZR, an iron chelator, can reduce anthracycline-induced cardiac damage, but other potent iron chelators, such as deferoxamine, deferiprone, and deferasirox, have yet to show this protective effect (Sultan et al., 2017).
3.3 Mitochondrial dysfunctionAs the primary site for cellular energy production and aerobic respiration, mitochondria are primarily composed of the mitochondrial outer membrane, intermembrane space, mitochondrial inner membrane, and mitochondrial matrix (Varga et al., 2015). In addition to their roles in energy conversion, the tricarboxylic acid cycle, oxidative phosphorylation, and calcium storage (Nunes-Nesi et al., 2013), mitochondria also play significant roles in processes such as cell proliferation, metabolism, and apoptosis (Su et al., 2023). An increasing body of research suggests that anthracycline drugs induce cardiac damage associated with mitochondrial dysfunction. When anthracycline drugs enter cardiac muscle cells, they alter mitochondrial membrane permeability, causing the release of cytochrome c (Cyt c) into the cytoplasm, subsequently triggering apoptosis. Additionally, anthracycline drugs can inhibit the activity of mitochondrial inner transcription enzymes, leading to ongoing oxidative stress in damaged mitochondria and exacerbating apoptosis (Parker et al., 2001) (Figure 4).
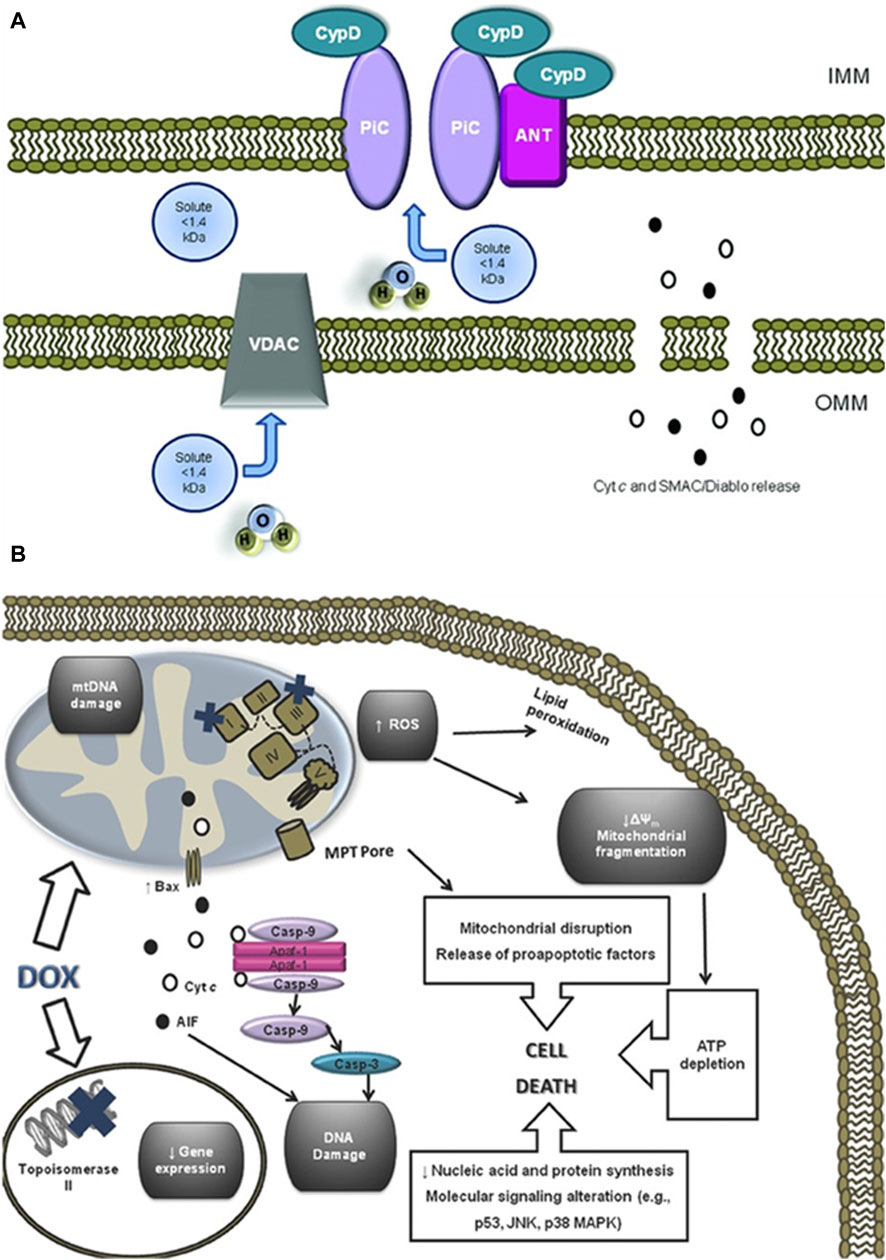
Figure 4. Anthracycline drugs induced changes in mitochondrial membrane permeability and induced cell death. (A) Mitochondrial permeability transition pore (MPTP) structure. It was recently proposed that the MPTP is generally considered to be a complex channel composed of several proteins, including voltage-dependent anion channel 1 (VDAC1) in the outer membrane, adenine nucleotide translocase 1 (ANT1) in the inner membrane, and cyclophilin D (CYPD) in the mitochondrial matrix. In addition, the MPTP can be regulated by other components, such as hexokinase (HK), creatine kinase (CK), and peripheral-type benzodiazepine receptors (PBR). Both antiapoptotic and proapoptotic members of the Bcl-2 family modulate the activity of MPTP (antiapoptotic members of the Bcl-2 family, including Bcl-2 and Bcl-XL, inhibit pore openingwhile proapoptotic Bcl-2 family members, such as Bax, Bak, and Bid, can induce MPT pore opening). Also, MPT pore opening can be inhibited by CypD ligands, such as cyclosporin A (CsA). The opening of MPTP leads to a collapse of transmembrane mitochondrial transmembrane potential and favors the release of apoptogenic proteins, such as cytochrome c (Cyt c). (B) A large component of DOXinduced cardiotoxicity is mediated by a redox cycle on mitochondrial complex I. Increased ROS generation by DOX redox cycle has several negative consequences, such as mitochondrial transmembrane potential disruption, MPTP formation, ATP depletion, and peroxidation of cellular membranes. Marked mitochondrial morphological disturbances induced by DOX include cristae disruption, matrix disorganization, and mitochondrial fragmentation. MPTP -induced outer membrane rupture due to osmotic swelling or permeabilization of the mitochondrial outer membrane mediated by proapoptotic proteins including BAX can lead to the release of cyt c and AIF. DOX also interferes with topoisomerase II, inhibiting DNA replication and preventing the repair of damage DNA strands. Finally, persistent downregulation of gene expression can be another consequence of DOX toxicity. All of these events may lead to cell death.
3.3.1 Mitochondrial membrane permeabilityCardiolipin is an essential component of the mitochondrial inner membrane. Anthracycline drugs exhibit a strong affinity for cardiolipin, forming irreversible complexes through positive and negative charge interactions and accumulating on the mitochondrial inner membrane (Parker et al., 2001). When the complex accumulates to levels exceeding 50–100 uM in the mitochondria, it leads to a significant increase in mitochondrial reactive ROS (Sarvazyan, 1996). High levels of ROS can result in oxidative reactions in mitochondrial proteins and lipids, leading to mitochondrial dysfunction and changes in mitochondrial membrane potential. Studies have shown that a decrease in mitochondrial membrane potential is present in almost all apoptotic cells and that this alteration occurs before the morphology of apoptotic cells changes, and the decrease in membrane potential is mainly due to the mitochondrial membrane permeability The decrease in membrane potential is mainly due to the permeability of the mitochondrial membrane (Zaib et al., 2022). Mitochondrial membrane permeability is regulated by the mitochondrial permeability transition pore (MPTP), which is located between the inner and outer mitochondrial membranes. It is generally considered to be a complex channel composed of several proteins, including voltage-dependent anion channel 1 (VDAC1) in the outer membrane, adenine nucleotide translocase 1 (ANT1) in the inner membrane, and cyclophilin D (CYPD) in the mitochondrial matrix.
VDAC1 is primarily located in the outer mitochondrial membrane of eukaryotic organisms, with a molecular weight of 35 KDa. In its open state, it can mediate the passage of ions and small molecules such as nucleotides (Benz et al., 1992). VDAC1 can also mediate the entry of respiratory chain substrates such as pyruvate, succinate, and malate into the mitochondria (Shoshan-Barmatz et al., 2010). VDAC1 is closely associated with the development of many diseases. Research has shown that VDAC1 is highly expressed in cancer cells (Shoshan-Barmatz et al., 2015). Arif et al. silenced VDAC1 expression by means of interfering RNA (siRNA) in cancer cells and found that siVDAC1 reduced VDAC1 levels by approximately 90% in lung cancer cells A549 and H358, prostate cancer cells PC-3, colon cancer cells HCT116, glioblastoma U87, hepatocellular carcinoma cells HepG2, and pancreatic cancer cells Panc-1 when the siVDAC1 was only at nanomolar concentrations. After transfection for 144 h, VDAC1 silencing persisted and significantly inhibited the growth of cancer cells. The mechanism of action is that when VDAC1 is downregulated in tumor cells, it leads to a decrease in mitochondrial membrane potential and adenosine triphosphate (ATP) levels, disrupting the exchange of substances between mitochondria and the cytoplasm, thereby inducing apoptosis. Furthermore, in a mouse model of lung cancer, intraperitoneal injection of si-VDAC1 not only inhibited tumor growth but also resulted in tumor disappearance. Therefore, this study suggests that inhibiting the overexpression of VDAC1 could be a novel therapeutic target for cancer treatment (Arif et al., 2017). Meanwhile, studies have reported that VDAC1 is also involved in the development of diabetic nephropathy. Gong et al. replicated a rat model of diabetic nephropathy using streptozotocin injections, and the results showed that VDAC1 expression significantly increased in the renal tissues of the model group, along with the occurrence of oxidative stress and severe interstitial nephritis (Gong et al., 2009). In an in vitro experiment of diabetic cardiomyopathy (DCM), it was found that when cardiomyocytes were transfected with lncRNA H19, the expression of miR-675 inside the cells decreased. Luciferase reporter gene experiments showed that miR-675 intertargets with VDAC1. When miR-675 inhibitor was transfected, VDAC1 expression increased, promoting cell apoptosis. When lncRNA H19 was highly expressed, it reduced the expression of VDAC1 inside the cells, inhibiting the occurrence of cardiomyocyte apoptosis. This experimental result suggests that the lncRNA H19/miR-675 axis regulates apoptosis by targeting VDAC1 (Li X. et al., 2016). Xu et al. found that in an experiment inducing apoptosis of mouse foot cell by DOX, the IP3R/glucose-regulated protein 75 (Grp75)/VDAC1/mitochondrial calcium uniporter (MCU) signaling pathway was activated, resulting in Ca2+ overload in mitochondria and an increase in cleaved caspase-3 levels, leading to apoptosis (Xu et al., 2018). No relevant research on the association between anthracycline drug-induced heart damage and VDAC1 was found in the literature.
ANT1 is located in the mitochondrial inner membrane, with a molecular weight of 33 kDa, and it accounts for 10% of the total mitochondrial proteins (Clémençon et al., 2013; Palmieri, 2013; Di Noia and Cirigliano, 2014). ANT1 is the sole ATP/adenosine diphosphate (ADP) transporter in eukaryotic cells, translocates back and forth between the inner mitochondrial membrane and the mitochondrial matrix to perform a translocation function, with its substrates being deoxyadenosine diphosphate (dADP), ADP, and ATP (Mielke et al., 2014). A study have shown that the protective role of Cyclosporine-A (CsA) on lung I/R injury depends on the inhibition of MPTP and CytC release, suppression of the activation of mitochondrial apoptosis pathway and the expressions of apoptotic-related proteins, as well as the decreased expression levels of ANT1 and VDAC1 (Li et al., 2017). Many inflammatory diseases are associated with abnormal activation of the NLRP3 inflammasome. The Src homology two domain of protein tyrosine phosphatase 2 (SHP2) in the cytoplasm can be transferred to mitochondria when an inflammatory stimulus enters the cell, interacts with ANT1, and leads to its overexpression, resulting in excessive activation of the NLRP3 inflammasome and triggering an inflammatory response (Guo et al., 2017). In a study of inducing myocardial infarction in wild-type (WT) and heart-specific ANT1 transgenic (ANT1-TG) rats, it was found that overexpressing ANT1 significantly reduced the myocardial infarction area and increased the survival rate of infarcted rats. At the same time, it reduced the release of mitochondrial Cyt c and activation of caspase-3. These results suggest that overexpression of ANT1 can compensate for damaged ANT activity, inhibiting apoptosis (Klumpe et al., 2016). Previous research has confirmed the involvement of ANT1 in anthracycline drug-induced heart damage. Matthias et al. found that in mice with DOX-induced cardiac damage, the expression of ANT1 mRNA in cardiac tissues significantly increased, leading to energy transport blockage within mitochondria and causing apoptosis (Schwebe et al., 2015).
CYPD is located in the mitochondrial matrix, with a molecular weight of 20 KDa. As a member of the immunoaffinity family, CYPD plays a crucial role in protein folding in mitochondria and participates in mitochondrial immune suppression. Primary isolation of embryonic fibroblast MEF cells from CYPD knockout mice and subsequent stimulation with H2O2 revealed that MEF cells lacking CYPD were less sensitive to H2O2-induced apoptosis compared to WT MEF cells, suggesting that CYPD is involved in oxidative stress-induced cell death (Delbridge et al., 2011). Moreover, when CYPD-deficient mice were subjected to acute middle cerebral artery occlusion and reperfusion, it was found that the infarction area was significantly reduced compared to normal mice. At the same time, the levels of SOD and catalase (CAT) significantly increased, demonstrating the important role of CYPD in ischemic brain injury models (Schinzel et al., 2005). In an experiment inducing acute kidney injury by cisplatin, it was found that when proximal renal tubule cells were specifically deficient in CYPD, it reduced the occurrence of kidney injury. The mechanism behind this was that cisplatin could enter the cells and cause peroxisome proliferator-activated receptor alpha (PPARα) to bind to CYPD. This, in turn, inhibited PPARα nuclear translocation and transcription, leading to fatty acid oxidation and apoptosis. When CYPD was absent, PPARα nuclear expression and transcription activity were maintained, preventing fatty acid oxidation and intracellular lipid accumulation, thereby exerting a protective effect on cells (Jang et al., 2020). A wealth of research has confirmed the involvement of CYPD in the development of anthracycline drug-induced cardiac damage. Dhingra et al. found that in a model of DOX-induced primary rat cardiac cell damage, the expression of the CYPD gene and protein increased after DOX treatment. This led to a decrease in mitochondrial membrane permeability, calcium accumulation in the mitochondria, and cell apoptosis. Further investigation into the mechanism found that DOX could activate the nuclear transcription factor (NF-κB)/Bcl-2 interacting protein 3 (Bnip3) signaling pathway, leading to an increase in CYPD expression and apoptosis (Dhingra et al., 2020).
A large body of research has confirmed that Bcl-2 family proteins regulate MPTP (Orsi et al., 2017). The Bcl-2 family proteins can be categorized into anti-apoptotic proteins and pro-apoptotic proteins. Anti-apoptotic proteins include Bcl-2, Mcl-1, and Bcl-xL, while pro-apoptotic proteins include Bim, Bid, Bak, and Bax. Most Bcl-2 family proteins have a hydrophobic region at the C-terminus of their peptide chains, allowing them to be positioned on the membrane of organelles. Additionally, the peptide chains of Bcl-2 family proteins contain four conserved helical domains, namely, BH4, BH3, BH2, and BH1, arranged from N-terminus to C-terminus. Among them, anti-apoptotic proteins highly homologous to Bcl-2 contain all four structural domains, making them Bcl-2-like proteins. Furthermore, pro-apoptotic proteins Bax and Bak also contain all four structural domains. Proteins containing only the BH3 domain are referred to as “BH3-only” proteins and are often pro-apoptotic. Their BH3 domain can bind to the hydrophobic groove formed by the BH1, BH2, and BH3 domains of anti-apoptotic proteins, forming heterodimers that control cell survival and apoptosis (Delbridge et al., 2016).
When anthracycline drugs enter cells, they bind to VDAC1 through Bax, leading to the opening of MPTP, a decrease in membrane potential, and the promotion of apoptosis (Kumarswamy and Chandna, 2009). The primary mechanism by which Bax affects VDAC1 is by altering the structure of VDAC1, leading to the loss of voltage dependence and selectivity for ion permeation, resulting in the release of Cyt c from the inner membrane. Bax can also interact with ANT1, forming a channel in the lipid bilayer. When Bcl-2 binds to ANT1, it inhibits the formation of the Bax-ANT1 channel, preventing changes in mitochondrial membrane permeability (Kroemer et al., 2007). Experimental evidence has shown that CYPD binds to Bcl-2 in a specific manner, and overexpression of Bcl-2 can suppress truncated Bid (tBid)-induced Cyt c release. In cells with normal Bcl-2 expression, the addition of a CYPD inhibitor can restore tBid-induced Cyt c release. These results strongly suggest that CYPD can be regulated through Bcl-2 (Eliseev et al., 2009).
Changes in mitochondrial membrane permeability result in the release of Cyt c into the cytoplasm, initiating apoptosis. Cyt c binds to apoptotic protease activating factor-1 (Apaf-1) in the cytoplasm, forming an apoptosome. Subsequently, the apoptosome recruits caspase-9 through the caspase recruitment domain (CARD) and caspase-3 activation, ultimately leading to cell apoptosis (Buttke and Sandstrom, 1994). Therefore, the release of Cyt c serves as a pivotal point for apoptosis initiation and is an essential marker for detecting mitochondrial pathway-induced cell apoptosis.
3.3.2 Mitochondrial transcription enzymeAdditional studies have found that anthracycline drugs can also cause mitochondrial dysfunction by inhibiting the activity of mitochondrial transcription enzymes. These transcription enzymes mainly include Top1mt and mitochondrial transcription factors (Zhang et al., 2012), and mitochondrial transcription factors include peroxisome proliferator-activated receptor-gamma coactivator-1α and 1β (PGC1α and PGC1β), Nuclear respiratory factor-1 (NRF1), mitochondrial transcription factor A (TFAM) (Yin et al., 2018), and Tumor suppressor protein 53 (p53). Research has found that histone deacetylase Sirtuin1 (SIRT1) plays a protective role against anthracycline-induced cardiac toxicity and its mechanism involves SIRT1 activating PGC1α to exert its protective effects (Cappetta et al., 2016). PGC1α, in turn, regulates the expression of TFAM, which bridges nuclear and mitochondrial signals and induces mtDNA transcription (Vega et al., 2015). At the same time, TFAM, together with Top1mt, maintains the stability of mtDNA (Khiati et al., 2014). Anthracycline drugs also affect the expression of the p53 gene (Zhuang et al., 2012), increasing the expression of p53 protein in the cytoplasm, promoting its binding with mitochondrial protein Parkin, inhibiting the clearance of damaged mitochondria, leading to sustained oxidative stress in damaged mitochondria, and exacerbating apoptosis (Hoshino et al., 2013). P53 gene-deficient mice, when subjected to DOX treatment, show a significant increase in mitochondrial integrity in cardiac muscles and a reduction in apoptotic cardiac muscle cells. Therefore, p53 might be one of the reasons for the occurrence of acute cardiac dysfunction after DOX treatment (Zhu et al., 2009; Dhingra et al., 2014; Fang et al., 2019) (Figure 4).
3.4 Muscle fiber degradation and sarcoplasmic reticulum calcium homeostasis imbalanceAnthracycline drugs’ cardiac damage is related to muscle fiber degradation and disruption (Sawyer et al., 2002). In vertebrate skeletal muscle fibers, Titin plays a crucial role in the generation and stability of sarcomeres, serving as the assembling framework for muscle fibers (Granzier and Labeit, 2004). Calpain, a type of protease, can cause the degradation of Titin (Lim et al., 2004). When anthracycline drugs induce cardiac damage, it leads to the overexpression of Calpain within cardiac muscle cells, causing muscle fiber disruption and necrosis (Ali et al., 2010). Storr and others have demonstrated that endogenous inhibitor of Calpain, Calpastatin, can alleviate anthracycline drug-induced cardiac toxicity (Storr et al., 2011). It’s worth noting that Titin degradation is an early process of cardiac cell damage triggered by DOX (Lim et al., 2004). Previous research has found Titin fragments in the urine of anthracycline-treated patients, which holds diagnostic significance for cardiac injury, reflecting potential myocardial damage (Tanihata et al., 2019) or skeletal muscle damage (Misaka et al., 2019). Therefore, Titin fragments can serve as a biomarker for detecting early anthracycline-induced cardiac damage.
Treatment with anthracycline drugs can cause an imbalance in intracellular sarcoplasmic reticulum calcium homeostasis (Sala et al., 2020). In normal cardiac muscle cells, Ca2+ is mainly stored in the mitochondria, sarcoplasmic reticulum, and sarcolemma, playing a vital role in maintaining the excitation-contraction coupling of cardiac muscle cells. Anthracycline drugs can disrupt the membrane’s permeability, activate Ca2+ channels in the sarcoplasmic reticulum, increase the release of Ca2+ from the sarcoplasmic reticulum to the cytoplasm, rapidly increase intracellular free Ca2+ concentration, affecting cardiac electrical activity, resulting in various arrhythmias, known as sarcoplasmic reticulum calcium overload. During anthracycline drug treatment, Ca2+/calmodulin-dependent protein kinase II (CaMKII) causes changes in the calcium signaling pathway-related genes (Luo et al., 2013). Impaired calcium homeostasis leads to the generation of intracellular reactive ROS, and anthracycline drugs reduce sodium and calcium exchange in the sarcolemma (Caroni et al., 1981). This calcium overload results in mitochondrial damage, aggravated oxidative stress, structural damage, and cell apoptosis. Anthracycline drugs can also inhibit the gene expression of Ca2+-ATPase on the sarcolemma of cardiac muscle cells, affecting the biosynthesis of Ca2+-ATPase, reducing its activity, reducing the sarcolemma’s Ca2+ uptake capability, leading to reduced ATP production in mitochondria, disrupting cardiac energy metabolism, worsening cell damage, and even causing cardiac cell death.
Anthracycline drugs can also activate cardiac matrix metalloproteinase 2 (MMP2) (Chan et al., 2018). In normal cardiac muscle cells, MMP2 is present in the sarcomeres and the cell cytoskeleton. When oxidative stress occurs, MMP2 is activated and responsible for degrading specific proteins, including Troponin I (TNI) (Wang et al., 2002), Myosin Light Chain 1 (MYL1) (Sawicki et al., 2005), α-Actinin (Sung et al., 2007), and Titin (Ali et al., 2010). Therefore, transgenic mice overexpressing MMP2 show reduced muscle contractile function, sarcomere disruption, and myosin protein hydrolysis (Bergman et al., 2007). In anthracycline drug-induced cardiac toxicity models, the increase in MMP2 levels and activity is a primary cause of early intracellular Titin protein hydrolysis and extracellular matrix remodeling (Chan et al., 2021). These adverse reactions can be prevented by oral MMP2 inhibitors, demonstrating the potential benefits of MMP2 inhibitors in preventing anthracycline-induced cardiac toxicity (Chan et al., 2021).
3.5 Oxidative stressMitochondria are the primary targets of anthracycline drugs’ actions. Cardiac muscle cells are rich in mitochondria, with 35%–40% more mitochondria compared to other cells, making them susceptible to damage (Goffart et al., 2004). In normal signal transduction processes, low levels of ROS are generated to maintain intracellular signaling, but under many pathological conditions, ROS levels increase from low to high (Liou and Storz, 2010; Peoples et al., 2019; Songbo et al., 2019). When anthracycline drugs enter cardiac muscle cells, they generate a significant amount of ROS by Generation of large amounts of ROS by reducing redox cycling in electron transport chain complex I, leading to ATP synthesis impairment (Davies et al., 1983). After the substantial ROS generation within mitochondria, anthracycline drugs transform into semiquinone radicals. Semiquinone radicals readily react with oxygen to generate O2-, which can react with SOD to produce H2O2. Simultaneously, anthracycline drugs also produce a substantial amount of iron ions within cardiac muscle cells. Iron ions, serving as the electron donors for H2O2 in the Fenton reaction, lead to the generation of -OH, sustaining ROS production (Canzoneri and Oyelere, 2008). Generated ROS react with mitochondrial lipids, proteins, and nucleic acids, disrupting mitochondrial functions, including protein translation, expression, and inducing lipid peroxidation, generating a substantial amount of malondialdehyde (MDA). MDA is one of the major products of membrane lipid peroxidation, and its content is often used to indicate the degree of cellular membrane lipid peroxidation. Studies have found that if intracellular ROS levels continuously increase, it can worsen cellular membrane lipid peroxidation, leading to an increase in MDA content (Liu et al., 2018). Therefore, MDA content can also indirectly reflect the extent of myocardial damage (Eder and Arriaga, 2006). SOD is a metalloenzyme that plays a crucial role in the oxidative and antioxidative balance of the body, It catalyzes the disproportionation of O2- to oxygen and hydrogen peroxide. (Henning et al., 2022).SOD can intercept the pathways of ROS-induced cell damage, timely repairing cell damage caused by ROS. As the primary scavenger of ROS within mitochondria, SOD plays an essential role in maintaining mitochondrial ROS levels and the stability of the mitochondrial environment (Bauer et al., 2014). In experiments on DOX-induced primary cardiac cell damage, DOX was found to increase intracellular MDA levels and reduce SOD activity, resulting in oxidative stress in cardiac cells (Dhingra et al., 2020). Wang et al. research showed that THP increased ROS levels in H9c2 cells and significantly elevated MDA content in rat serum while reducing SOD activity. RUT can reverse the oxidative stress damage caused by THP (Wang et al., 2018). In an experiment conducted by Aleixo and others on DOX-induced cardiac damage in rats, it was found that SOD activity decreased, MDA content increased in the cardiac tissues of model group rats compared to normal group rats, leading to the accumulation of a large amount of lipid peroxidation products in cardiac tissues, causing severe oxidative stress reactions (Marques-Aleixo et al., 2016) (Figure 5).
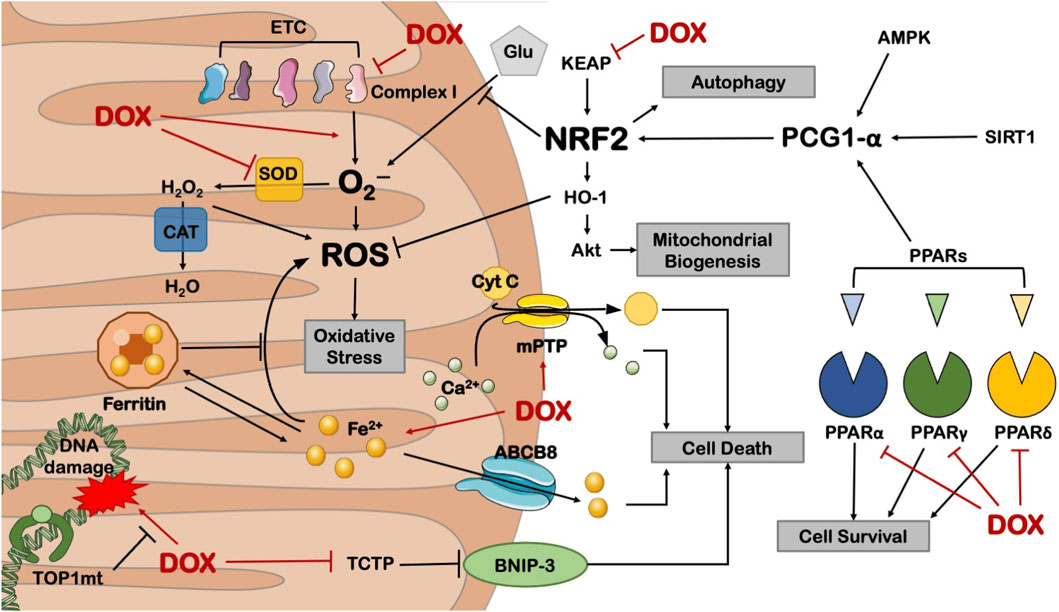
Figure 5. Anthracycline drugs can cause mitochondrial dysfunction by inhibiting the activity of mitochondrial transcription enzymes. These transcription enzymes mainly include Top1mt and mitochondrial transcription factors, and mitochondrial transcription factors include peroxisome proliferator-activated receptor-gamma coactivator-1α and 1β, Nuclear respiratory factor-1, mitochondrial transcription factor A (TFAM), and Tumor suppressor protein 53.
Research has also found that Nrf2, as a basic leucine zipper bZIP protein, can mitigate cell damage caused by ROS and electrophiles by regulating the expression of antioxidant proteins, keeping cells in a stable state, maintaining the dynamic balance of redox in the body, and counteracting oxidative damage. Nrf2 deficiency can exacerbate cardiac toxicity and cardiac dysfunction. In Nrf2 gene knockout (Nrf2−/−) mice, the adverse reactions of DOX cause cardiac muscle cell necrosis and cardiac dysfunction, which are related to oxidative stress, impaired autophagy, and increased accumulation of polyubiquitin aggregates (Li et al., 2014).
3.6 Cell apoptosisIn the normal physiological state, the cardiomyocyte is a terminally differentiated, non-proliferative mature cell. Therefore, apoptosis of cardiac myocytes plays a crucial role in the cardiotoxicity induced by anthracyclines. Recent studies have confirmed that anthracycline drugs can induce apoptosis in cardiac myocytes, thereby damaging the myocardium. The primary pathway of anthracycline-induced cell apoptosis is mitochondria-mediated intrinsic signaling. When anthracycline drugs enter cardiac myocytes, they cause the release of Cyt c from the mitochondrial inner membrane, which then binds to Apaf-1 to form the apoptosome. The apoptosome activates the caspase family, leading to cell apoptosis. AKT, as a key factor in the apoptosis signaling pathway, can inhibit cell apoptosis by regulating the Bcl-2 family. In addition to the mitochondria-mediated intrinsic signaling pathway, anthracycline drugs can also induce programmed necrosis and cell pyroptosis, resulting in damage to cardiac myocytes (Figure 6).
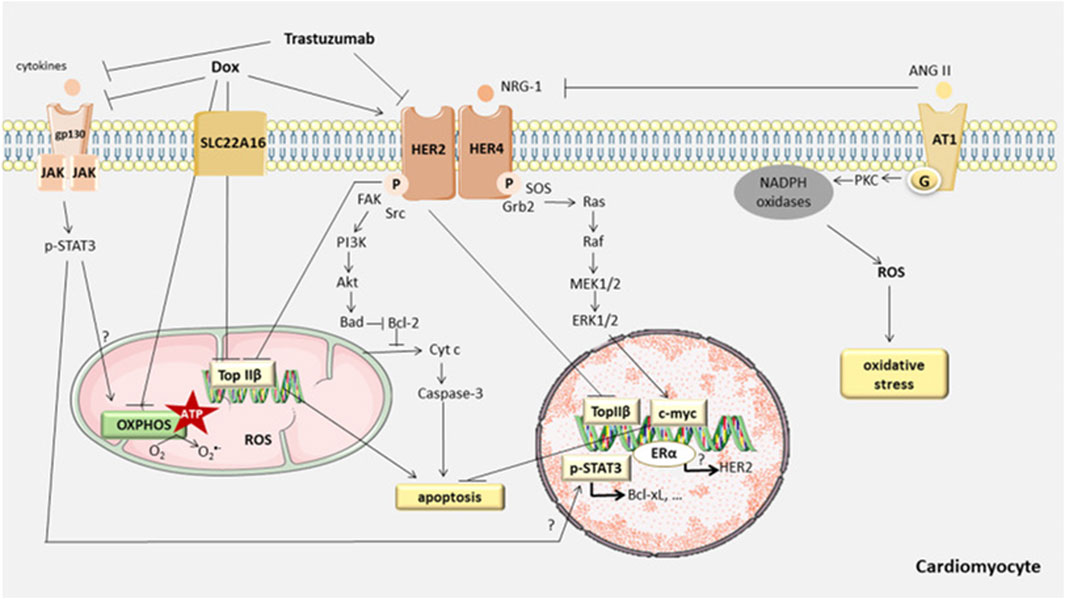
Figure 6. Anthracycline-induced cell apoptosis is mitochondria-mediated intrinsic signaling. When anthracycline drugs enter cardiac myocytes, they cause the release of Cyt c from the mitochondrial inner membrane, which then binds to Apaf-1 to form the apoptosome. The apoptosome activates the caspase family, leading to cell apoptosis. AKT, as a key factor in the apoptosis signaling pathway, can inhibit cell apoptosis by regulating the Bcl-2 family. In addition to the mitochondria-mediated intrinsic signaling pathway, anthracycline drugs can also induce programmed necrosis and cell pyroptosis, resulting in damage to cardiac myocytes.
3.6.1 Mitochondria-mediated intrinsic pathwayCell apoptosis (Apoptosis) is the primary mechanism of anthracycline anticancer activity and a major cause of non-cancer cell toxicity, especially in non-regenerative organs like the heart. Cell apoptosis primarily involves the regulation of two signaling pathways: the extrinsic pathway, which relies on cell surface “death” receptors, and the intrinsic pathway, which relies on mitochondria (Chowdhury et al., 2006). When oxidative stress or damage occurs within the cell, the mitochondria-mediated intrinsic signaling pathway is activated, leading to cell apoptosis. The mitochondria-mediated intrinsic cell apoptosis pathway primarily depends on the balance of pro-apoptotic and anti-apoptotic factors within the cell, which are members of the Bcl-2 superfamily. These factors control the permeability of the mitochondrial membrane (Skommer et al., 2007). The mitochondrial inner and outer membranes contain factors that can initiate the apoptotic cascade, such as Cyt c. When the permeability of the mitochondrial membrane is altered after anthracycline entry into the cell, Cyt c is released into the cytosol, where it can bind to Apaf-1 (Jourdain and Martinou, 2009), generating apoptotic vesicles, which can aggregate cysteoaspartic enzymes and activate cyst caspases through proteolytic cleavage, resulting in apoptotic cell death. (Soengas et al., 1999). The Bcl-2 family can be divided into three subtypes: Bcl-2-like anti-apoptotic factors, Bax-like pro-apoptotic factors, and BH3-only protein pro-apoptotic factors (Ferrans, 1978). Research has shown that serine/threonine kinase (AKT) can directly regulate the Bcl-2 family and indirectly regulate cell apoptosis by modulating apoptosis-related transcription factors. AKT, also known as protein kinase B (PKB), is a serine/threonine protein kinase with a molecular weight of approximately 60 kDa. AKT can be divided into three types (AKT1, AKT2, AKT3, or PKBα, PKBβ, and PKBγ), each with distinct functions but with some overlap. AKT1 is widely distributed in tissues, AKT2 is mainly found in muscle and fat cells, while AKT3 is expressed in the testes and the brain. AKT regulates various biological processes, including cell apoptosis, proliferation, growth, and glucose metabolism. The AKT signaling pathway is closely associated with the development of various diseases, such as malignancies, diabetes, and heart diseases (Franke et al., 1997; Linton et al., 2019; Uko et al., 2020).
Studies have reported that AKT inhibits cell apoptosis by primarily suppressing the activity of related apoptosis factors. BH3-only proteins (BOPs), as pro-apoptotic factors, can promote the binding of Bad to Bcl-2 or Bcl-xl. Bad regulates its binding to Bcl-2 through phosphorylation sites. When not stimulated by growth factors, Bad remains non-phosphorylated. Non-phosphorylated Bad consistently binds to Bcl-2, preventing Bcl-2 from exerting its anti-apoptotic function. When AKT is activated, it phosphorylates Bad at serine 136 (Nechushtan et al., 1999), causing Bad to dissociate from the Bcl-2 complex and bind to the scaffolding protein 14-3–3 (Masters et al., 2004). Binding of Bad to 14-three to three proteins initially isolates it from the mitochondrial outer membrane and prevents dephosphorylation of Bad, thus inhibiting its re-association with Bcl-2, which affects the cell-protective function of Bcl-2.
AKT can also directly regulate another member of the Bcl-2 family, Bax. Bax is a pro-apoptotic factor that can exert its function only when located in the mitochondrial outer membrane. In the absence of apoptosis stimuli, Bax is found in the cytoplasm. When cell apoptosis occurs, Bax undergoes conformational changes, enabling it to aggregate and insert into the mitochondrial outer membrane more easily (Gardai et al., 2004). Once inserted into the mitochondrial outer membrane, Bax forms oligomeric pores, allowing Cyt c and other pro-apoptotic factors to be released from the mitochondrial membrane. AKT phosphorylates Bax in a manner similar to Bad (Xin and Deng, 2005). AKT phosphorylates Bax at Ser184, inhibiting its conformational changes, thus preventing Bax from translocating to the mitochondrial membrane (Yamaguchi and Wang, 2001). This prevents the formation of oligomeric pores and the release of pro-apoptotic factors, thereby inhibiting cell apoptosis. Previous studies have found that in rat models of heart damage induced by anthracycline drugs, DOX induces apoptosis in cardiac tissues by inhibiting the AKT/Bcl-2 signaling pathway (Zhang Y. et al., 2020). Bai et al. made similar observations in a mouse model of DOX-induced cardiac damage, demonstrating that DOX can reduce p-AKT and Bcl-2/Bax protein expression, leading to the occurrence of cell apoptosis (Bai and Wang, 2019).
3.6.2 Other pathwaysAKT can also promote the expression of Fibronectin type III domain-containing protein 5 (FNDC5), which possesses certain cardioprotective properties. FNDC5 can inhibit the degradation of nuclear factor-erythroid2 related factor2 (Nrf2), mitigating anthracycline-induced cardiac damage (Zhang X. et al., 2020). Nrf2 plays a role in protecting myocardial cells by stabilizing the redox homeostasis and autophagic flux (Zhao et al., 2018).
In addition to apoptosis, anthracyclines induce other forms of cell death, including programmed necrosis (Necroptosis). Research has confirmed the association of necroptosis with the Ca2+/CaMKII pathway (Zhang T. et al., 2016) and the BH3 region proteins BOPs/Adenovirus E1B 19 kDa Interacting Protein3 (BNIP3) signaling pathway (Dhingra et al., 2014). However, inhibiting cell apoptosis or necroptosis only partially enhances myocardial cell survival (Zhang T. et al., 2016), indicating the involvement of other signaling pathways, such as pyroptosis (Meng et al., 2019). Pyroptosis results in cell swelling until the cell membrane ruptures, leading to the release of cellular contents and activating a strong inflammatory response.
4 Genetically driven mechanisms of anthracycline cardiotoxicityIdentifying patients at high risk of anthracycline-induced cardiac injury using guidelines and predictive models remains extremely challenging. In addition, available parameters are not sufficient to explain why only some high-risk patients develop cardiotoxicity after treatment with antitumor drugs. The dose of chemotherapy with anthracyclines that results in toxic reactions varies significantly between patients. For example, some patients are able to tolerate a dose of 1,000 mg/m2 of adriamycin, whereas others develop acute cardiotoxicity at a dose of 200 mg/m2. These phenomena suggest that there may not be a “safe” anthracycline dose for all patients. This suggests that sensitivity to anthracyclines varies significantly among individuals and reveals the importance of genetic drivers of anthracycline-induced cardiotoxicity (Mulrooney et al., 2009) (Figure 7).
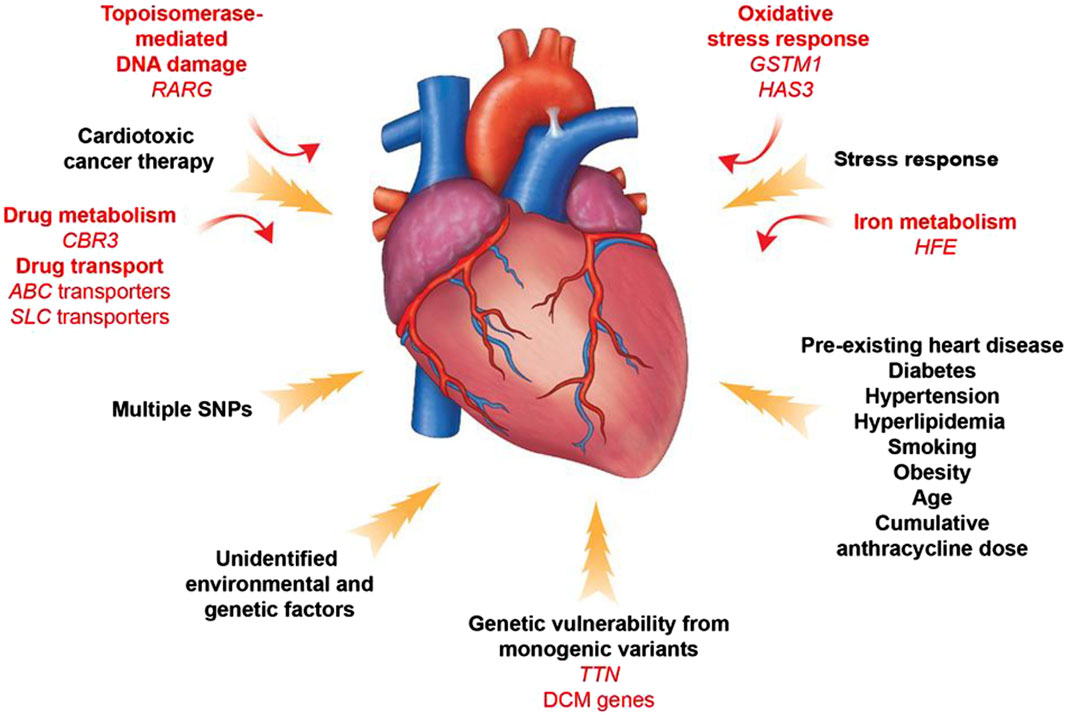
Figure 7. Contributors to Anthracycline -induced cardiovascular toxicity. A combination of clinical and genetic risk factors leads to increased risk of developing toxicity upon cancer therapy treatment. Elucidation of genetic contributors of cancer therapy-induced cardiovascular toxicity facilitates understanding of its molecular mechanism and development of its therapeutic strategies.
In a genome-wide unbiased chain-directed association study based on the HapMap project online CEU dataset, a total of 31,312 high-frequency single-nucleotide polymorphisms (SNPs) spanning over 1,278 genes were examined, as well as 86 lymphoblastoid cell lines susceptible to zorubicin. It was found that the heritability of the IC50 value for Zoerythromycin was 0.29. (Duan et al., 2007). Pathway analysis showed that phosphatidylinositol signaling system, GPI-anchored proteins, and axon guidance pathway were frequent in the list of candidate genes. This study proposes that the cardiotoxicity caused by Zoerythromycin may be influenced by many genes, with each gene having an impact at varying amounts.
In a large-scale candidate gene study involving 1,697 subjects, researchers chose genes related to the metabolism of reactive oxygen species, drug transport and metabolism, DNA repair, endothelial physiology, the renin-angiotensin-aldosterone system, muscle contraction and structure, inflammation, and cell cycle (Wojnowski et al., 2005). The genotypes of polymorphisms in 73 genes were determined in peripheral blood cells of individuals with non-Hodgkin’s lymphoma. These genotypes were then analyzed to determine if they were associated with acute and chronic cardiotoxicity. The authors concluded that anthracycline-induced cardiotoxicity was significantly associated with genetic polymorphisms in NAD(P)H oxidase and efflux transporter proteins (MRP1 and MRP2). An extensive review conducted a thorough analysis of 28 papers investigating the association between potential genes, analyzing a total of 84 genes and 147 single nucleotide polymorphisms (Leong et al., 2017). The presence of three risk alleles in the ABCC2, CyBA, and RAC2 genes was found to dramatically elevate the likelihood of developing anthracycline-induced cardiotoxicity.
Another experiment aimed to identify common variants that transmit disease risk in the general population. Researchers used a genotyping array to study patient DNA samples, which contained SNPs near candidate genes or whole genome SNPs. When using the latter approach, genome-wide association studies (GWAS) incorporate estimation by statistically analyzing the haplotypes in a genotyped reference panel to infer SNPs that are not directly genotyped. By far the most common group of genes associated with cardiovascular toxicity are involved in drug transport. The adenosine triphosphate-binding cassette transporter (ABC) proteins are dynamic cellular transporters for several medicines, including anthracyclines (Januchowski et al., 2014). Polymorphisms in specific genes, such as ABCB4, ABCC1, and ABCC2, have been linked to a higher likelihood of developing heart problems after receiving anthracycline treatments in both children and adults with blood-related and solid tumors (Wojnowski et al., 20
留言 (0)