
記住我




Atherosclerosis is one of the prominent pathological conditions that lead to myocardial infarction and stroke. A significant risk factor for its development is chronological age (1), accelerated by epigenetic aging (2). Aging is associated with declines in cellular homeostasis causing inflammation (3–6) and organ system dysfunction (7–9), thereby resulting in the distortion of the healthy balance between inflammation and tissue homeostasis (10). Aging affects this balance in multiple ways including decreased efficiency of cellular housekeeping and tissue repair functions (11–13), which leads to the accumulation of cellular debris (14), increased endoplasmic reticulum stress (15) defective tissue homeostasis (16, 17), declining endocrine functions (18) changes in RNA compositions (19) immune senescence (20, 21), and increased inflammation (22, 23). Understanding how interactions between these systems can affect the outcomes of pharmacological studies (24), offering opportunities to slow the effects of aging (25) and prevent the development of associated diseases (26–28).
The central actors in maintaining tissue homeostasis are macrophages (29, 30), dendritic cells (31, 32), stem cells (33), and vitamin D (34–38). However, our understanding of their interaction remains incomplete, as evidenced by multiple inconclusive clinical trials seeking to halt the progression of cardiovascular disease associated with vitamin D deficiency (39–43). In previous reports, we have reported that vitamin D deficiency affects the tipping points that control network–network interactions in over 500 diseases, including atherosclerosis (44).
Materials and methods Background informationCause–effect relationships in complex biological network systems are determined using an information theory-based analysis methodology that is developed to ascertain information flows induced by drugs and diseases (probes) in biological networks (45). This cause–effect relationship-driven analysis approach identifies global network architectures mediating probe-induced information transfers within and between tissues and ascertains how probes affect network linkage (46–54). The statistical value of solutions resulting from this analysis is extremely significant because the global network architecture discovered in system-wide cause–effect analysis links information in large, orthogonal, and decentralized data sets. Furthermore, no single data point can distort the global network architecture imposed by the functional and structural interdependence of subunits linked in cause–effect relationships.
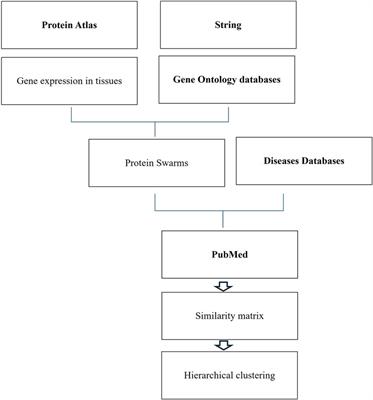
Protein swarm generation and spectral clusteringFunctional analysis of genomic and non-genomic signaling by vitamin D in 4,821 diseases as illustrated in Figure 1 involved use of (1) the data mining algorithm developed by SystaMedic Inc. in collaboration with the University of Connecticut, (2) the databases and data analysis tools of the STRING platform (55), (3) tissue-associated protein expression data identified in the Human Protein Atlas (56), (4) information published in Medline (57), and (5) data analysis and visualization tools of Spotfire (58). Hierarchical clustering of similarity matrices obtained through Medline data extraction was used for identifying global network architectures linking probe-induced information flows in tissue–protein networks and cause–effect relationships (59–63).
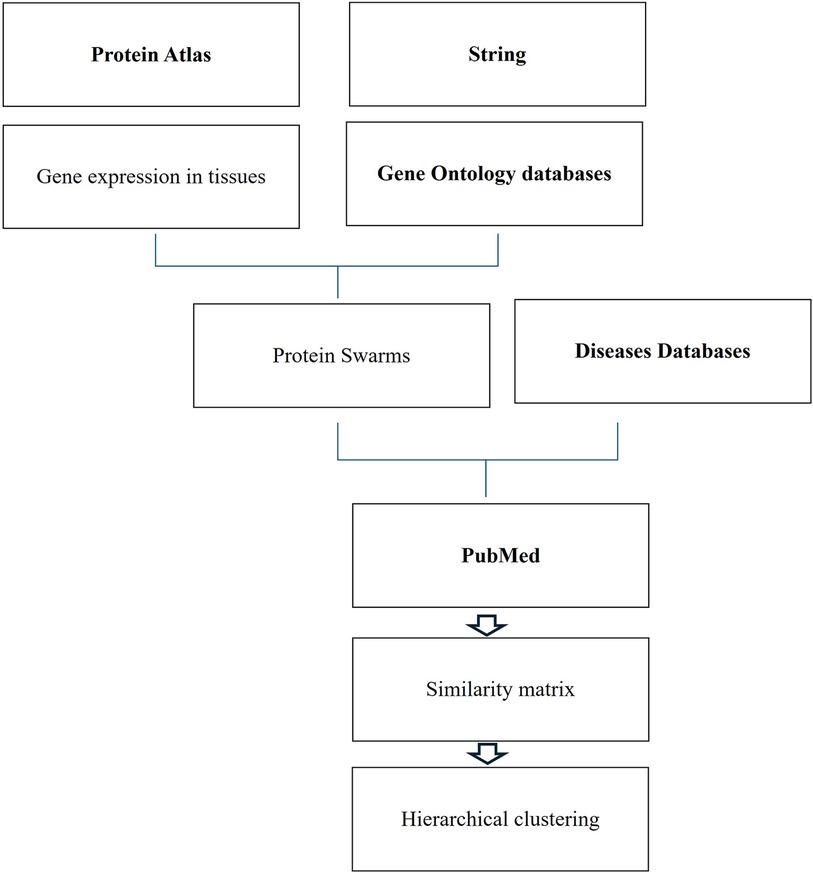
Figure 1 Databases and work flow used for cause effect analysis.
Tissue-specific protein expression data (tissue proteomes) extracted from the Human Protein Atlas are processed using gene enrichment analysis in the STRING platform. The output of this analysis allows the identification of tissue-associated protein network fragments that contain no more than five proteins capable of interacting in a swarm-like fashion with other proteins in response to probe-induced system perturbations. Henceforth, the term protein swarms in lieu of protein network fragments is used in this analysis. Identifying and collecting protein swarms representing all tissues in the Human Protein Atlas provides descriptor sets that can be used for determining co-citation frequencies of probes and protein swarms in Medline. Viewing co-citation frequencies measurements as estimates of a probe's capacity to affect information transfers within and between protein swarms, hierarchical clustering of similarity matrices obtained through Medline data extraction identifies global network architectures linking probe-induced information flows in tissue–protein networks and cause–effect relationships.,
TerminologyThe term vitamin D herein refers to the hormone 1,25-dihydroxyvitamin D3 (calcitriol) and not its precursor, calcidiol, which is commonly used for measuring vitamin D deficiencies (64). In turn, genomic and non-genomic signaling refers to the catenation of functions resulting from (1) the binding of calcitriol at picomolar concentrations to the nuclear vitamin D receptor (VDR) (65) and (2) the binding of calcitriol at nanomolar concentrations to the membrane receptor PDIA3, also known as ERp57 and grp58 (66, 67). PDIA3 is an endoplasmic reticulum chaperone with protein disulfide isomerase activity. It regulates vitamin D genomic signaling (68); apoptosis (69, 70); efferocytosis (71), mitochondrial functions (72); functions of macrophages (73), dendritic cells (74, 75), and stem cells (76); progranulin (77); and functions of the innate and adaptive immune system (78).
Results Establishment of a global effect positioning systemIdentification of co-citation frequencies of 4,821 diseases with 7,371 protein swarms in Medline, construction of a 4,821 × 7,371 similarity matrix, and UPGMA hierarchical clustering of the similarity matrix using cosine as similarity measure were used to identify and select swarm clusters linked within a confidence in cluster similarity value (CCSV)of >0.92 with swarms containing vitamin D receptors (79). This process identified 29 discrete clusters containing in aggregate 656 protein swarms holding 363 proteins (disease-associated vitamin D interactome; Table 1).
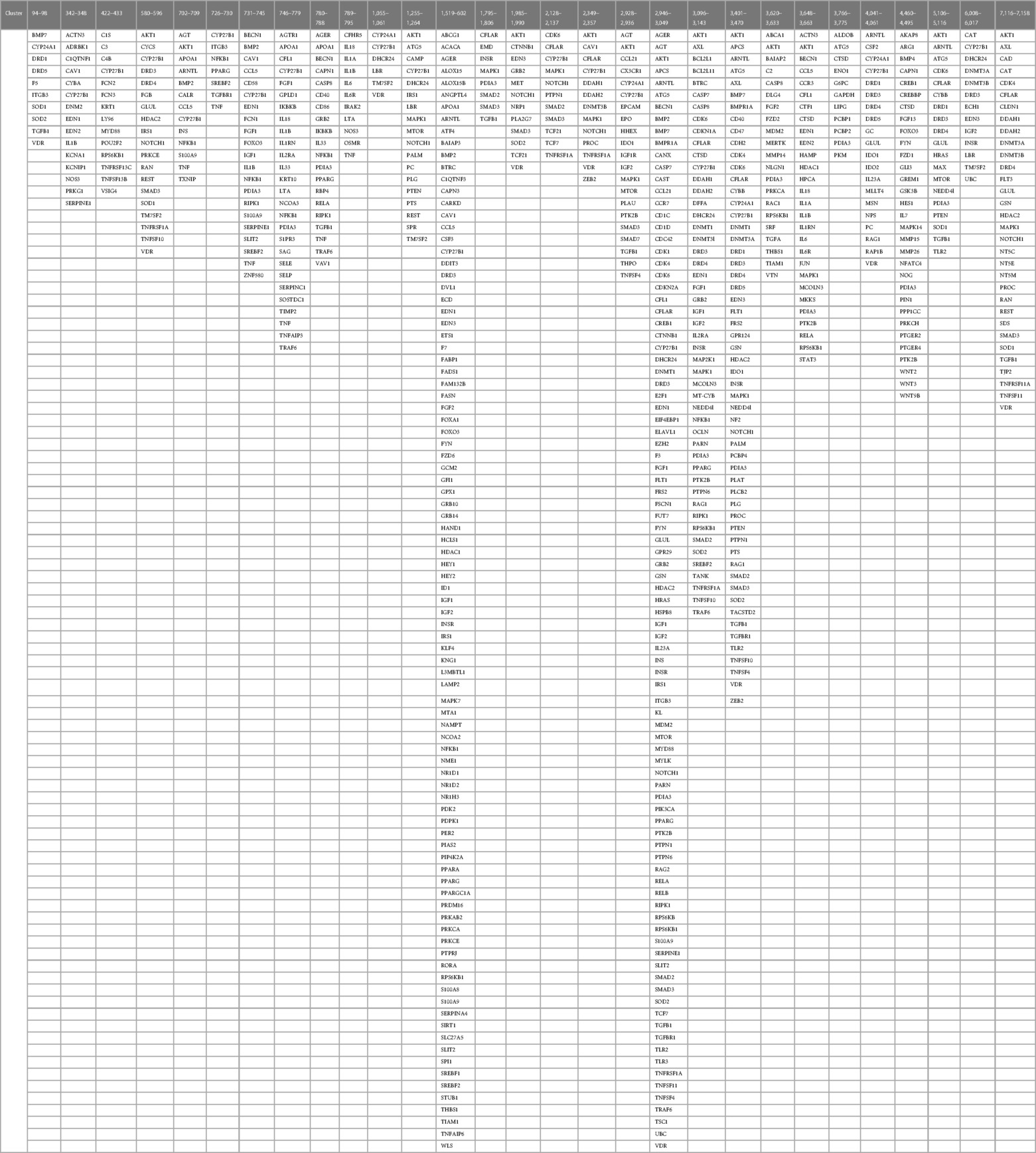
Table 1 The proteins regulating vitamin D's genomic and non-genomic pharmacologies in 4,821 diseases isolated from 29 discrete swarm clusters containing in aggregate 363 proteins (disease-associated vitamin D interactome).
Relevance assessment of the vitamin D interactome in diseaseFigure 2 shows a protein network constructed from 363 proteins that were identified in 656 protein swarms described in Table 1. Further analysis to determine known disease associations of these proteins showed that 234 out of the 363 proteins in the vitamin D interactome have statistically significant disease associations (p-value 2.06 × 10−33). These findings establish the linkage between vitamin D signaling and the 4,821 diseases studied

Figure 2 The physical interactions between 363 proteins captured in 29 vitamin D receptors containing swarm clusters. A total of 234 proteins (red) have known disease associations.
Determination of the role of vitamin D signaling in 4,821 diseases and identification of cause–effect relationships associated with cardiovascular disease phenotypes
Vitamin D receptor-associated protein swarms were labeled according to cluster membership and used for collecting co-citation frequencies associated with 4,821 diseases in a second round of Medline data mining. Hierarchical clustering of the resulting 4,821 × 656 similarity matrix followed by data visualization provided the heatmap shown in Figure 3.
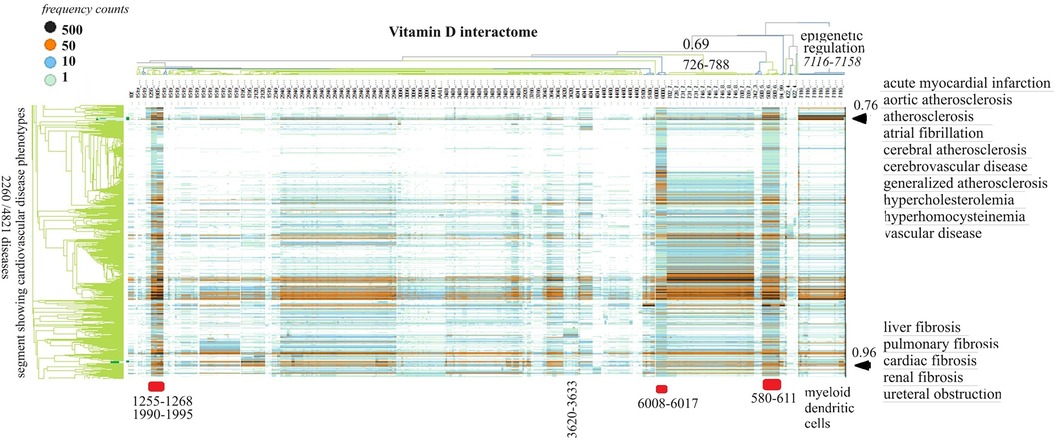
Figure 3 The horizontal dendrogram identifies the clusters of protein swarms that contain proteins engaged in similar functions. The vertical axis identifies the groups of diseases with similar cause–effect relationships. The hierarchical organization of protein swarms (CCSV > 0.69) shows that vitamin D-regulated processes are tightly coupled and subject to epigenetic regulation involving functions of myeloid dendritic cells (cluster 7,116–7,158).
Inspection of co-citation frequencies associated with disease and swarm clusters suggests that proteins in swarm clusters 580–611, 1,255–1,268, 1,990–1,995, and 6,008–6,017 (underscored in red) play a prominent role in 2,260 diseases. Furthermore, the dominant position of swarm cluster 7,117–7,158, in the hierarchical organization of protein swarms, indicates that vitamin D-mediated signaling is subject to epigenetic regulation involving myeloid dendritic cells (80, 81). Protein network construction using proteins in clusters 7,116–7,158 followed by functional analysis identified that protein interactions in these clusters regulate nitric oxide signaling (82), redox signaling (83), DNA methylation (84), and histone deacetylation (85). Similarly, the evaluation of cause–effect relationship similarities between diseases revealed that atherosclerosis, acute myocardial infarction, atrial fibrillation, cerebrovascular disease, hypercholesterolemia, hyperhomocysteinemia, and vascular disease are subject to similar regulatory mechanisms involving swarm clusters 580–611, 1,255–1,268, 1,990–1,995, 6,008–6,017, and 7,116–7,158 (CCSV > 0.76) (29).
Vitamin D signaling in cardiovascular diseasesThe effects of vitamin D signaling in cardiovascular diseases were further examined in three separate categories: (1) genomic signaling, (2) a mixture of genomic and non-genomic, and (3) non-genomic signaling.
Vitamin D genomic signalingThe initial focus was to determine functions regulated by proteins in swarms 7,117–7,158, 580–611, 1,255–1,268, 1,990–1,995, and 6,008–6,017 involved in 2,260 diseases containing the nuclear vitamin D receptor. Network construction using proteins in these clusters and retrieval of 10,000 publications with statistically significant protein network overlaps identified cardiovascular disease-associated functions regulated by the nuclear vitamin D receptor. Network overlaps between these functions are shown in Table 2. The results indicate that vitamin D's genomic signaling regulates protein–protein interactions involved in atherosclerosis, efferocytosis (B) (86, 87), macro-autophagy (C) (88), macrophage autophagy (D) (89, 90), macrophage polarization (E) (91, 92), cellular senescence (F) (93), ER stress response (G) (94), tissue homeostasis (H) (95), innate immunity (I) (96). caveola-mediated endocytosis (J) (97), and apoptosis (K) (98).
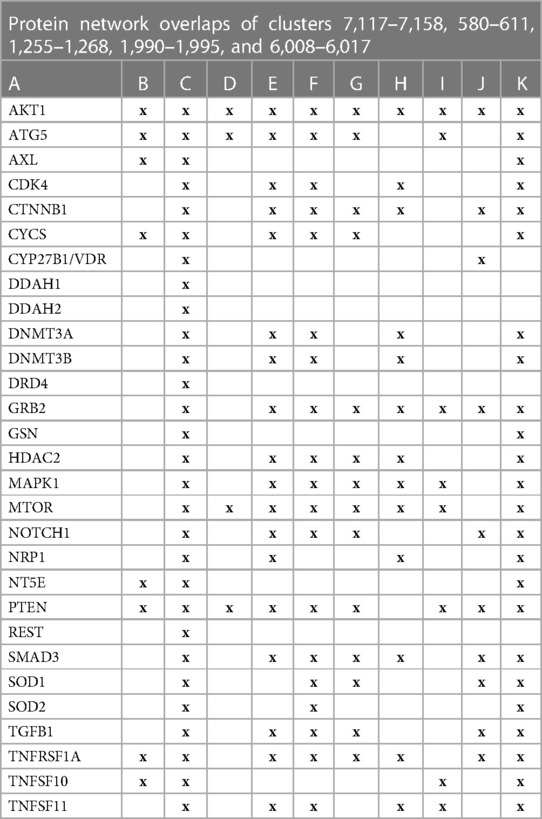
Table 2 The interactions between proteins in swarm clusters 7,117–7,158, 580–611, 1,255–1,268, 1,990–1,995, and 6,008–6,017 and overlaps of protein networks associated with atherosclerosis (A), efferocytosis (B), macro-autophagy (C), macrophage autophagy (D), macrophage polarization (E), cellular senescence (F), ER stress response (G), tissue homeostasis (H), innate immunity (I), caveola-mediated endocytosis (J), and apoptosis (K).
A protein network-centered view of results shown in Table 2 demonstrates that genomic signaling of vitamin D regulates at least 10 different functions involved in atherosclerosis (99–102) (Figure 4). Moreover, extensive protein network overlaps between these functions suggest that vitamin D's genomic signaling modulates a monitoring scheme that responds to nitric oxide signaling (DDAH1, DDAH2) (103), redox signaling (SOD1, SOD2, PDIA3) (104), environmental signals (DNMT3A, DNMT3B) (105, 106), histone acetylation (HDAC2) (107), inflammatory signals (TNFRSF1A, TNFSF10) (108), growth factor signaling (TGFB1, GRB2, NRP1) (109–111), mitochondrial status (CYS) (112), metabolic changes (MTOR) (113), and calcium signaling (GLSN) (114). Reflecting the importance of macrophages in atherosclerosis, Figure 4 shows that this regulatory scheme integrates macrophage polarization (115) and macrophage autophagy (88).
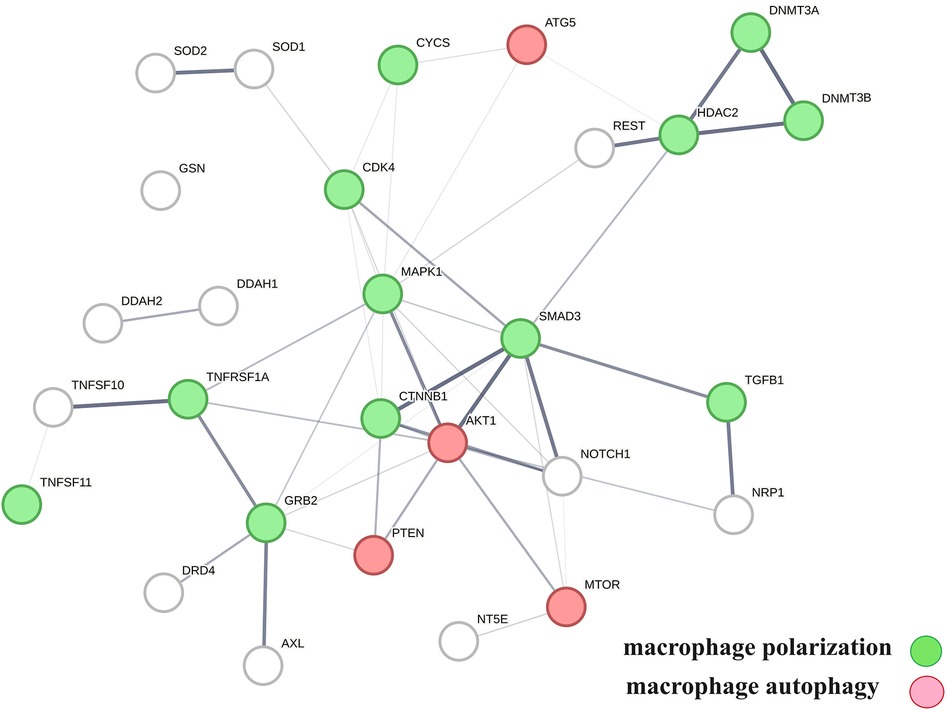
Figure 4 The physical interactions between proteins in swarm clusters 7,117–7,158, 580–611, 1,255–1,268, 1,990–1,995, and 6,008–6,017, highlighting the overlaps between protein networks associated with atherosclerosis (no color), macrophage polarization (green), and macrophage autophagy (red). Proteins ATG5, AKT1, and PTEN are involved in the regulation of all functions.
Vitamin D genomic and non-genomic signalingTo evaluate how the interaction between vitamin D's genomic and non-genomic signaling affects atherosclerosis, we focused on swarm cluster 726–788 (CCSV 0.99) containing the nuclear and non-nuclear vitamin D receptors. Protein network construction and retrieval of publications with statistically significant protein network overlaps identified cardiovascular disease-associated functions (Table 3).
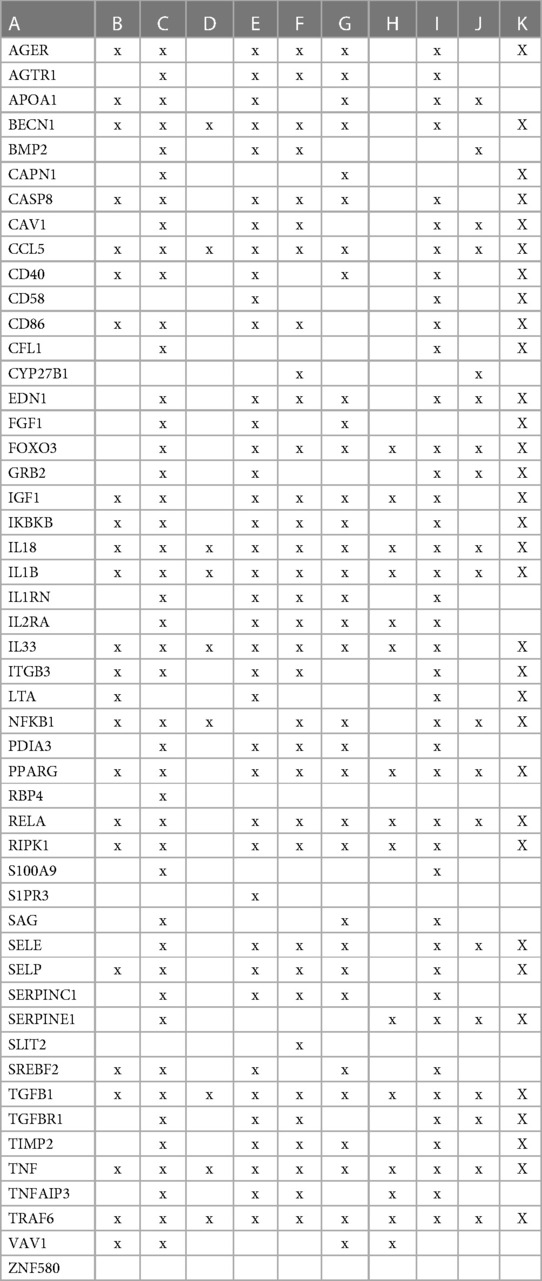
Table 3 The interactions between proteins in swarm cluster 726–788 and overlaps of protein networks associated with atherosclerosis (A), efferocytosis (B), macro-autophagy (C), macrophage autophagy (D), macrophage polarization (E), cellular senescence (F), ER stress response (G), tissue homeostasis (H), innate immunity (I), caveola-mediated endocytosis (J), and apoptosis (K).
A protein network-centered view of Table 3 is shown in Figure 5. It highlights the interactions between proteins regulating macrophage polarization and macrophage autophagy in atherosclerosis (21). Specifically, the proteins colored in green regulate macrophage polarization, and the proteins highlighted in red are constituents of the NF-κB complex, which regulates inflammatory responses (116), autophagy, and macrophage autophagy (117). IL18 and TRAF6 nodes in the macrophage autophagy network regulate inflammatory processes and plaque instability in atherosclerosis (118, 119). Thus, this indicates that a mix of genomic and non-genomic signaling is involved in the regulation of atherosclerotic plaque stability.
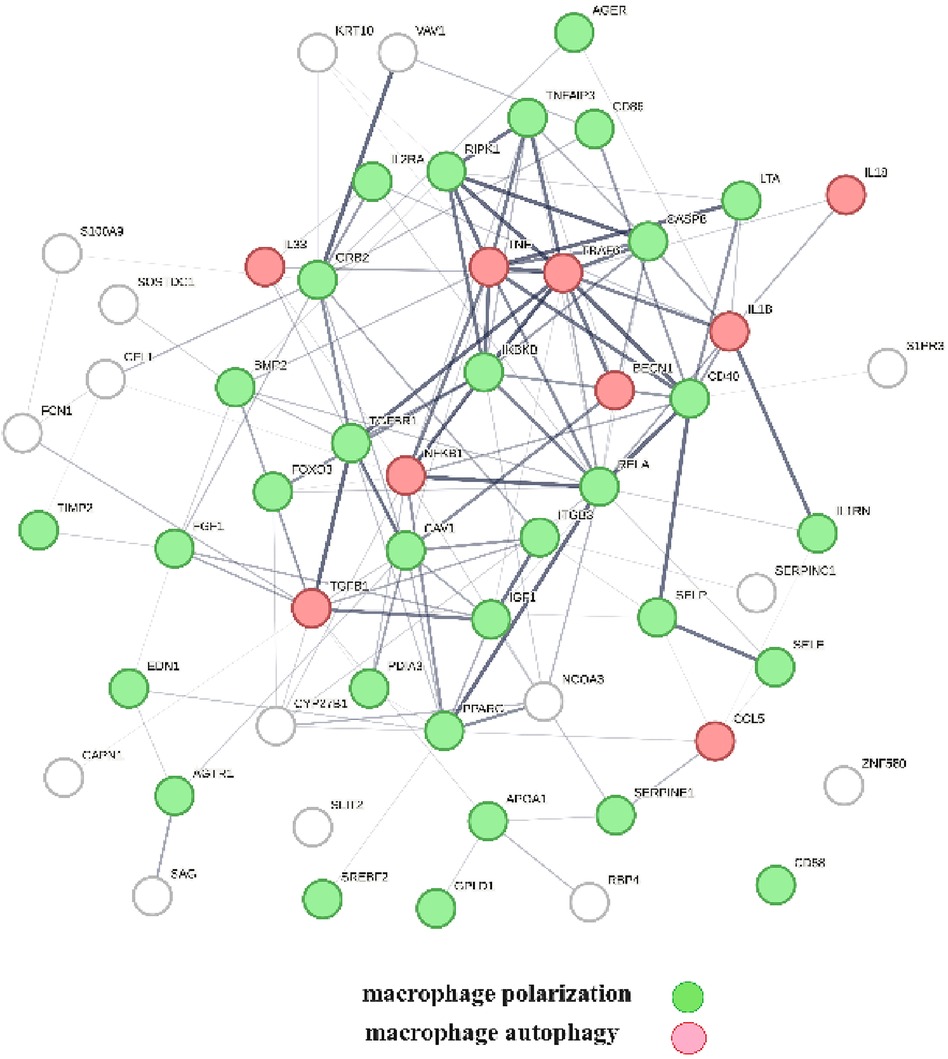
Figure 5 The protein interactions regulating vitamin D's genomic and non-genomic signaling in atherosclerosis. Proteins colored in green regulate macrophage polarization, and proteins colored in red regulate macrophage autophagy.
Vitamin D non-genomic signalingThe first step prior to conducting cause–effect analysis was to identify clusters that only contained the non-nuclear vitamin D receptors. Specifically, there are clusters 3,780–788, 1,795–1,806, 3,620–3,633, 3,648–3,663, 3,766–3,775, 4,460–4,495, and 5,106–5,116. The next step was to evaluate if proteins in these clusters could physically interact with vitamin D's non-genomic receptor, i.e., PDIA3. This analysis identified that PDIA3 can physically interact with AKT1, BAIAP2, ENO1, GAPDH, GSK3B, MTOR, NFKB1, PCBP2, PCBP2, PDIA3, PPARG, SOD1, STAT3, and TGFB1. Protein network construction using this group of proteins and retrieval of publications with statistically significant network overlap identified that the interactions between these proteins modulate PI3K/AKT/mTOR signaling and that the natural product rutin, targeting this regulatory scheme, displays a U-shaped (non-linear) dose–response relationship in cardiomyocytes (120).
The involvement of the PDIA3 interactome in atherosclerosis was further assessed by identifying potential network–network interactions regulated by proteins in swarm cluster 3,620–3,633 (CCSV0.99) containing PDIA3 and proteins expressed in all tissues. Protein network construction using proteins in 3,620–3,633 clusters and retrieval of publications with statistically significant protein network overlaps identified cardiovascular disease-associated functions shown in Table 4. In contrast to the observations in Tables 2, 3, the absence of proteins regulating macrophage autophagy in Table 4 suggests that vitamin D's genomic and non-genomic signaling play different roles in regulating autophagy.
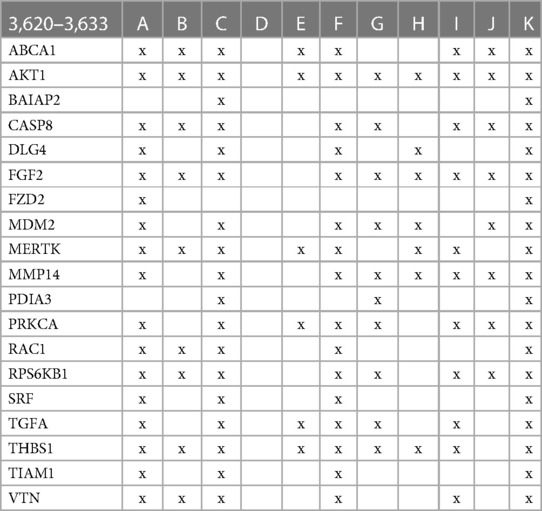
Table 4 The interactions between proteins in swarm cluster 3,620–3,633 and overlaps of protein networks associated with atherosclerosis (A), efferocytosis (B), macro-autophagy (C), macrophage autophagy (D), macrophage polarization (E), cellular senescence (F), ER stress response (G), tissue homeostasis (H), innate immunity (I), caveola-mediated endocytosis (J), and apoptosis (K).
Figure 6 provides a protein network-centered view of Table 4 and shows that proteins regulating vitamin D's non-genomic pharmacology can physically interact and affect atherosclerosis by regulating the balance between autophagy (121), apoptosis (122), endoplasmic reticulum stress response (94), cellular senescence (123, 124), and tissue homeostasis (125).
A key finding based on analysis of protein interactions in this network is that PDIA3 interactome (Figure 6) plays a key role in atherosclerosis. For example, interactions of PDIA3 with AKT1 (126) and interactions of AKT1with TIAM1 regulate the expression of the multicellular protein osteopontin (127, 128), which is involved in generating the atheromatous pathology of atherosclerosis (129). Furthermore, interactions of PDIA3 with brain-specific angiogenesis inhibitor 1-associated protein 2 (BAIAP2; a.k.a. IRSP53) regulate apoptotic cell clearance (130), plasma membrane shape homeostasis, and endocytosis. Mutations in tyrosine kinase-defective Mertk receptor MERTK decreases macrophage autophagy and increases the size of necrotic plaque in atherosclerosis (131, 132). Interactions of BAIAP2 with TIAM1, RAC1, and DLG4 regulate filopodia extension via actin polymerization (133) and regulate LDL metabolism by macrophages (134, 135). In addition, interactions of BAIAP2 with the WAVE regulatory complex (136, 137) control highly dynamic processes involved in cholesterol clearance, embryogenesis, neuron morphogenesis and plasticity, immune cell activation, chemotaxis, fibrosis, and cancer invasion and metastasis (138–141).
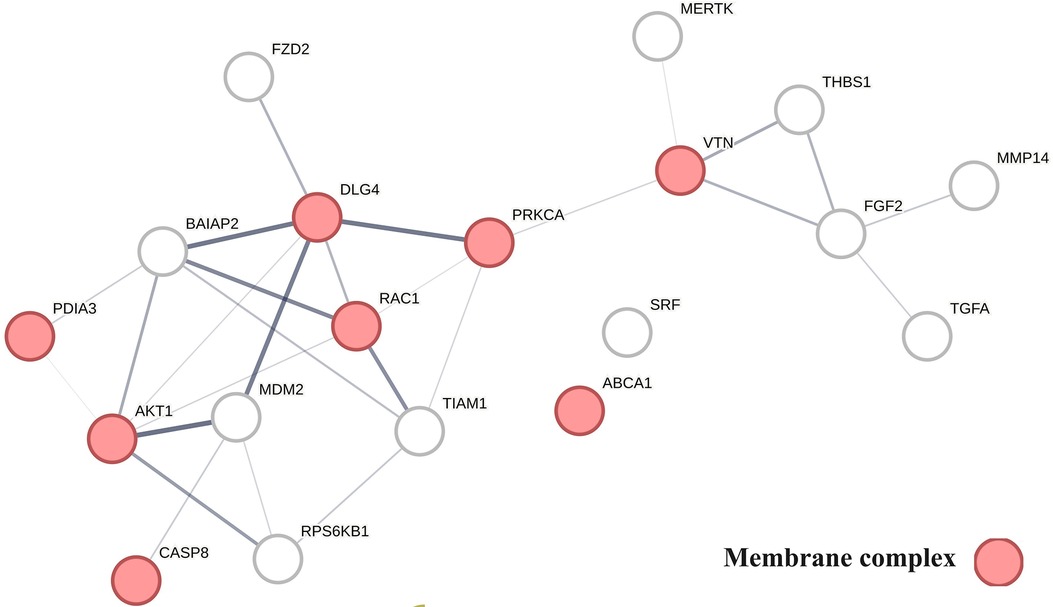
Figure 6 The physical interactions between proteins regulating vitamin D's non-genomic pharmacology in atherosclerosis.
The finding that PDIA3 interactome regulates multiple reciprocal feedback loops is supported by observations showing that calcitriol, in combination with p53 aka TP53 regulates the expression of MDM2 (142), occupying a central position as shown in Figure 6. Further, the regulation of MDM2 expression creates an autoregulatory feedback loop wherein the expression of MDM2 is coregulated by TP53 protein levels and MDM2 protein levels decrease the ability of TP53 to function as a positive transcription factor for MDM2 (143). Since the regulation of TP53 expression regulates the inactivation of SREBP2, this feedback loop regulates the activities of mevalonate and PCSK9 pathways, which are key players in atherosclerosis (144, 145). Similarly, reciprocal regulation of TP53 expression and nitric oxide levels via regulation of expression of nitric oxide synthases (NOS2, NOS3) (146, 147) and methylation and demethylation of arginine (swarm cluster 7,116–7,158) creates additional feedback loops (148, 149). Closing these feedback loops, MDM2 regulates the expression of the nuclear vitamin D receptor (150, 151), and calcitriol, in coordination with TGFB1, regulates PDIA3 expression (152, 153). Thus, these two feedback loops regulate the ratio of nuclear and non-nuclear expression of vitamin D receptors. This finding suggests that this form of regulation can fine-tune the ratio between genomic and non-genomic signaling of vitamin D and can operate independently from the binding affinities of calcitriol. Since vitamin D effects are antifibrotic whereas PDIA3 effects are profibrotic, these feedback loops may be involved in balancing processes involved in tissue homeostasis.
Adding further complexity to the TP53-mediated regulatory scheme is that ER stress-induced posttranslational modifications of P53 affect the sensitivity of MDM2 and TP53 feedback loops (154, 155). The impact of these feedback loops on the outcomes of pharmacological studies using vitamin D is manifested in clinical and preclinical observations showing U-shaped dose–response relationships (156–160).
DiscussionThis study provides deeper insights into the role of vitamin D in cardiovascular disease development. The use of a spectral clustering method for identifying vitamin D-regulated processes in 4,821 diseases provides a global effect positioning system that can be used to determine cause–effect relationships in atherosclerosis. Examination of vitamin D-associated genomic and non-genomic pharmacologies using this positioning system revealed origins of non-linear dose–response relationships observed in preclinical and clinical studies with vitamin D. In addition, it also identified functional relationships between multiple reciprocal feedback loops that sometimes regulate the opposing functions activated by nuclear and membrane-associated vitamin D receptors. Among the processes of relevance for the treatment of cardiovascular diseases are p53-mediated regulation of atherosclerotic plaque stability and macrophage autophagy, which are known to safeguard the plasticity of macrophages in tissue homeostasis. We have used an unsupervised machine learning approach for deciphering these complex cause–effect relationships and used protein network analysis for identifying interactions between protein networks that regulated the functions that are of relevance in atherosclerosis. The initial step was aimed at establishing a global effect positioning system (Table 1) using information theory-based methodology to allow for an unbiased analysis of vitamin D pharmacology in diseases. Information flow-based cause–effect analysis of 4,821 diseases led to the identification of the vitamin D interactome (Figure 2) comprising 363 proteins, where 64% of proteins were found to have highly statistically significant (p-value = 2.06 × 10−33) disease associations. This observation establishes the value of our protein swarm-based framework for investigating cause–effect relationships in protein–protein interaction (PPI) networks and complements previous work reported by Silverbush and Sharan (161) who used drug response and cancer genomic data to orient the human PPI network.
Examination of the role of the vitamin D interactome in 4,821 diseases provided two key findings: (1) vitamin D-regulated processes are involved in over 2,000 diseases including cardiovascular disease, and (2) all functions regulated by vitamin D are subject to epigenetic regulation (Figure 3). Epigenetic regulation of atherosclerosis is well established (162), and our finding of the involvement of vitamin D signaling contributes to the development of diagnostics or therapeutic agents in atherosclerosis. Also depicted in Figure 3 is the observation that cardiovascular diseases including atherosclerosis are affected by PPI in swarms 580–611, 1,255–1,268, 1,990–1,995, 6,008–6,017, and 7,116–7,158.
The effects of vitamin D signaling in cardiovascular diseases were further examined as three separate categories: (1) genomic signaling mediated by PPI in swarms 580–611, 1,255–1,268, 1,990–1,995, 6,008–6,017, and 7,116–7,158 (Table 2), (2) mixture of genomic and non-genomic signaling mediated by PPI in swarms 726–788 (Table 3), and (3) non-genomic signaling mediated by PPI in swarms 3,620–3,633 (Table 4). Tables 2–4 show the results of evidence-based cause–effect analysis using network overlaps of proteins in the three swarm categories with PPI networks of atherosclerosis and the 10 biological functions affected by aging and involved in atherosclerosis. The significance of vitamin D's genomic, mixed genomic, and non-genomic signaling in atherosclerosis related to the 10 biological functions associated with atherosclerosis (Figures 4–6) was determined by measuring the extent of network overlap, ability to engage in physical interactions, and centrality of shared network nodes. Hence, the main inference from the results shown in Tables 2, 3 and Figures 4, 5 is that genomic and mixed genomic signaling by vitamin D modulates the 10 important biological functions associated with atherosclerosis. Additionally, examination of network overlaps between these 10 biological functions indicated that they are part of an integrated regulatory scheme that includes macrophage polarization and macrophage autophagy as central elements.
The effects of vitamin D's non-genomic signaling are summarized in Table 4 and Figure 6. The absence of overlap between proteins in swarm cluster 3,620–3,633 and macrophage autophagy in combination with the observation that these clusters are enriched with proteins involved in fibrosis suggests that non-genomic signaling by vitamin D plays an important role in tissue homeostasis.
The identification of protein network–network interactions described above allows the generation of a comprehensive view detailing how interactions between the 10 biological functions affect atherosclerosis.
A critical factor in atherosclerosis-associated mortality is plaque stability (163), where stable plaque forms carry a low risk of sudden mortality while rupture of unstable forms causes myocardial infarction and stroke. Our analysis (Tables 2, 3; Figures 4, 5) indicates that vitamin D signaling plays a key role in balancing plaque stability (164) by regulating macrophage autophagy (90) and macrophage polarization, a key determinant of the M1/M2 ratio, where macrophages with M1-like characteristics are associated with the inflammatory stages of atherosclerosis while macrophages with M2-like characteristics have anti-inflammatory characteristics and promote plaque regression (91, 165). Declining macrophage autophagy increases macrophage apoptosis, decreases macrophage efferocytosis, increases oxidative stress, and destabilizes plaque (132, 166).
A unique property of macrophages is that they can directly generate calcitriol, which is the most active form of vitamin D, in response to a variety of input signals (167). Calcitriol, in turn, activates macrophage autophagy and shifts the macrophage polarization toward anti-inflammatory M2-like macrophages (168, 169). Thus, by shifting macrophage polarization toward M2 (170), calcitriol can decrease inflammation (171) and increase the clearance of cellular debris by efferocytosis (172).
Additionally, of relevance to the development of atherosclerosis is aging. Aging shifts macrophage polarization toward M1 phenotypes (173), increases cellular senescence (174) and inflammation (175), and decreases macrophage functions (176). Calcitriol can counter aging-induced acceleration of atherosclerosis at the plaque initiation (177–179), progression (179, 180), rupture (181), regression (182–184), and healing stages of atherosclerosis (179, 185). Accordingly, shifting macrophage ratios from M1 to M2 controls plaque size and stability (170). For example, low shear stress or high endoplasmic reticulum negatively affects autophagy, and shifting macrophage polarization toward inflammatory M1 accelerates atherosclerosis (186–190). Vitamin D deficiency exacerbates this development by increasing M1 macrophage populations and decreasing autophagy (115, 191). Calcitriol administration can reverse this trend by shifting macrophage populations toward M2 phenotypes, which increases macrophage autophagy (192–195).
However, an important finding of our investigation is that the over-enrichment of M2 macrophage populations can result in excessive fibrosis. In other words, vitamin D's non-genomic signaling via PDIA3 at higher calcitriol concentrations promotes fibrosis, leading potentially to plaque destabilization (196). Maintenance of the delicate balance between M1 and M2 macrophage subpopulations is critical as it affects tissue homeostasis. Experimental data suggest that too much or too little on either side of the equation results in detrimental outcomes (197). This balance can be affected in multiple ways. For example, the involvement of immune responses in this delicate balancing act is implicated by observations that cathelicidins produced by vitamin D's genomic response to infections activate autophagy and shift macrophage populations toward M2 (198). In addition, the identification of regulatory schemes clearly establishes that vitamin D regulates multiple feedback loops (76, 199–203) involved in tissue homeostasis, which are subject to additional regulation by nitric oxide (204) and redox signaling (205). Evidence of these types of non-linear (U-shaped) cause–effect relationships/dose responses, expected due to opposing pharmacologies, is indeed observed in numerous preclinical and clinical studies with vitamin D [see references (156–160)].
Strength and limitationThe dynamics of protein–protein interactions impart functional diversity to proteomes and balance functions separating health and diseases. From a whole organism perspective, single-cell approaches do not provide the system-level insight that is needed to understand how interactions between proteomes at tissue levels affect the progression of a disease or the responses of organisms to treatments (206). To generate the required insights, integrated approaches are needed that can determine relationships between large heterogeneous, unstructured, and noisy data stored in different databases. By taking advantage of the modular designs of proteomes and principles governing information transfers in integrated systems (207, 208), protein swarm-based cause–effect analysis delivers unbiased insight into how interactions between tissue- and cell-associated proteomes regulate the physiological and pathological states of organisms.
“Swarm Intelligence” based analysis reveals cause-effect relationship between the many variables involved and provides understanding that is very difficult to ascertain with other means.
The reach of the current vitamin D-centered cause–effect analysis is limited by its focus on determining the system pharmacology of calcitriol. Therefore, our study neglects the potential contributions of calcidiol or the various vitamin D metabolites. Furthermore, technical constraints limit the resolution of network–network interactions by setting upper limits to the number of protein swarms used for sampling tissue proteomes. Lastly, the limiting capability of machine learning approaches in the biological cause–effect analysis is the scarcity of data on the concentration time dependency of dynamic cause–effect relationships at a system-wide scale.
SummaryUsing a global framework for proteomics-based cause–effect analysis, our analysis identified a broad vitamin D-regulated scheme involved in tissue homeostasis of over 2,000 diseases. By providing a perspective on how interactions between vitamin D's genomic and non-genomic actions, nitric oxide and redox signaling, macrophage autophagy, macrophage polarization, and p53 signaling shape tissue homeostasis, our analysis addresses a long-standing enigma presented by observations that vitamin D supplementation, despite projected benefits, produces little benefits in clinical studies (209). Recognizing the importance of p53, nitric oxide, and redox signaling in vitamin D-regulated processes offers the opportunity to increase the efficacy of vitamin D supplementation in cardiovascular diseases. Toward this end, modalities integrating nitric oxide redox signaling and vitamin D-centered pharmacologies have already shown promising outcomes (210). Refinement of these approaches is expected to improve the treatment of cardiovascular diseases and increase the quality of life in aging (211).
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Materials; further inquiries can be directed to the corresponding author.
Author contributionsAF: Writing – original draft. SK: Writing – review & editing.
FundingThe authors declare that no financial support was received for the research, authorship, and/or publication of this article.
AcknowledgmentsRon Sostek contributed valuable feedback on use of Swarms intelligence in complex System analysis.
Conflict of interestAF and SK are founders of Emergent System Analytics LLC.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1388025/full#supplementary-material
References2. Sánchez-Cabo F, Fuster V, Silla-Castro JC, González G, Lorenzo-Vivas E, Alvarez R, et al. Subclinical atherosclerosis and accelerated epigenetic age mediated by inflammation: a multi-omics study. Eur Heart J. (2023) 44(29):2698–709. doi: 10.1093/eurheartj/ehad361
Crossref Full Text | Google Scholar
6. Gogulamudi VR, Durrant JR, Adeyemo AO, Ho HM, Walker AE, Lesniewski LA. Advancing age increases the size and severity of spontaneous atheromas in mouse models of atherosclerosis. Geroscience. (2023) 45(3):1913–31. doi: 10.1007/s11357-023-00776-8
PubMed Abstract | Crossref Full Text | Google Scholar
7. Boss GR, Seegmiller JE. Age-related physiological changes and their clinical significance. West J Med. (1981) 135(6):434–40. PMID: 7336713; PMCID: PMC12733167336713
PubMed Abstract | Google Scholar
8. Lv J, Zhang C, Liu X, Gu C, Liu Y, Gao Y, et al. An aging-related immune landscape in the hematopoietic immune system. Immun Ageing. (2024) 21(1):3. doi: 10.1186/s12979-023-00403-2
PubMed Abstract | Crossref Full Text | Google Scholar
9. Ambale-Venkatesh B, Liu CY, Liu YC, Donekal S, Ohyama Y, Sharma RK, et al. Association of myocardial fibrosis and cardiovascular events: the multi-ethnic study of atherosclerosis. Eur Heart J Cardiovasc Imaging. (2019) 20(2):168–76. doi: 10.1093/ehjci/jey140
PubMed Abstract | Crossref Full Text | Google Scholar
11. Hu H, Cheng X, Li F, Guan Z, Xu J, Wu D, et al. Defective efferocytosis by aged macrophages promotes STING signaling mediated inflammatory liver injury. Cell Death Discov. (2023) 9(1):236. doi: 10.1038/s41420-023-01497-9
PubMed Abstract | Crossref Full Text | Google Scholar
12. Zhong W, Rao Z, Xu J, Sun Y, Hu H, Wang P, et al. Defective mitophagy in aged macrophages promotes mitochondrial DNA cytosolic leakage to activate STING signaling during liver sterile inflammation. Aging Cell. (2022) 21(6):e13622. doi: 10.1111/acel.13622
PubMed Abstract | Crossref Full Text | Google Scholar
14. Gladyshev VN, Kritchevsky SB, Clarke SG, Cuervo AM, Fiehn O, de Magalhães JP, et al. Molecular damage in aging. Nat Aging. (2021) 1(12):1096–106. doi: 10.1038/s43587-021-00150-3
PubMed Abstract | Crossref Full Text | Google Scholar
15. Zhang Z, Guo Q, Zhao Z, Nie M, Shi Q, Li E, et al. DNMT3B activates FGFR3-mediated endoplasmic reticulum stress by regulating PTPN2 promoter methylation to promote the development of atherosclerosis. FASEB J. (2023) 37(8):e23085. doi: 10.1096/fj.202300665R
PubMed Abstract | Crossref Full Text | Google Scholar
16. Jones L, Passegue E. Stem cells homeostasis and aging. Innov Aging. (2023) 7(Suppl 1):259–60. doi: 10.1093/geroni/igad104.0862
Crossref Full Text | Google Scholar
18. van den Beld AW, Kaufman JM, Zillikens MC, Lamberts SWJ, Egan JM, van der Lely AJ. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. (2018) 6(8):647–58. doi: 10.1016/S2213-8587(18)30026-3
PubMed Abstract | Crossref Full Text | Google Scholar
20. Smit V, de Mol J, Schaftenaar FH, Depuydt MAC, Postel RJ, Smeets D, et al. Single-cell profiling reveals age-associated immunity in atherosclerosis. Cardiovasc Res. (2023) 119(15):2508–21. doi: 10.1093/cvr/cvad099
留言 (0)