
記住我




Inherited single amino acid substitutions are an important source of potential phenotypic variation between individuals that can lead to disease risk (1, 2) and can contribute to complex multifactorial disorders (3). About one-half of known genetic conditions are caused by nonsynonymous single nucleotide polymorphisms (nsSNPs) (3, 4). Single amino acid mutational studies are limited but naturally-occurring missense variants with associated phenotypes can provide very valuable information for analysis of structure-function relationships of proteins. Techniques such as targeted exome sequencing play key roles in discovering alleles associated with Mendelian and complex disorders.
A comprehensive review of disease-associated PLG missense variants in world populations is highly relevant to more fully comprehend their overall causative influences on coagulopathies and inflammatory diseases and the involvement of the fibrinolytic system in these processes. In this review, we explore the ramifications of naturally occurring variants on the abundant multi-functional soluble plasma protein zymogen, PLG, and its activated product, the serine protease, plasmin. We begin this review with a summary of the background on PLG/plasmin structure-function which is necessary to better understand the mechanisms of the effects of missense variants on the properties of PLG and plasmin.
Human plasminogenHuman plasminogen is encoded by the PLG gene, which is located on human chromosome 6q26. The PLG DNA contains 19 exons separated by 18 introns and is 51,861 bp in length (http://genome.ucsc.edu/) (5). PLG is translated primarily in the liver (6), along with minor production in extrahepatic cells. The translated protein is a single-chain 810 amino acid protein without enzymatic activity. Upon maturation, the 19-amino acid signal peptide is removed, and two carbohydrate chains are placed on PLG side-chains, Asn289 and Thr346 (Figure 1) (7–9), as well as a phosphorylation site of unknown significance located at Ser578 (10).
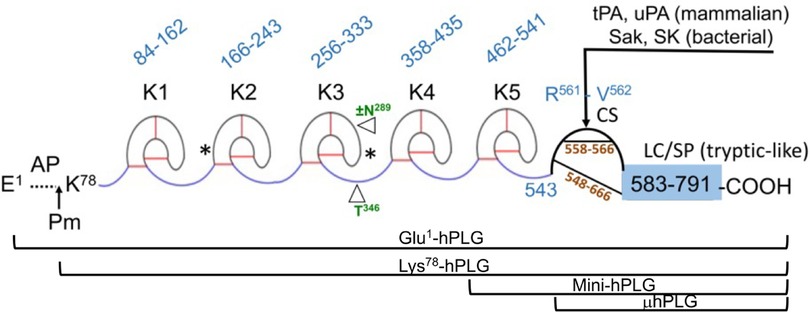
Figure 1 The mature form of the zymogen, human plasminogen (Glu1-PLG). After cleavage of the 19-amino acid residue signal sequence, the protein contains 791 amino acids in a single chain. A heavy chain (HC) of 561 amino acids is comprised of five ∼80 amino acid triply disulfide linked kringle (K) domains with inter kringle linker regions (ID). A 229-amino acid light chain (LC) is homologous to serine proteases (SP) such as trypsin. This protease chain is silent in intact PLG but becomes active when PLG activators (PA) catalyze cleavage of the Arg561-Val562 peptide bond at the cleavage site (CS), providing human plasmin with the LC doubly disulfide-linked to the HC at residues 558/566 and 548/666. The AP is released during this activation process by the generated plasmin. The final plasmin contains residues Lys78-Asn791 (Lys78-PLG) with the HC and LC linked by two disulfide bonds. Note that both the HC and the LC are latent in the zymogen. A single N-linked glycosylation site is present at Asn289, which is occupied in ∼60% of the mature protein molecules and a single O-linked glycosylation site at Thr346 is occupied in 100% of the mature protein molecules. Other post-translational forms of PLG that occasionally appear in the literature are bracketed below the Figure.
In numbering PLG residues, the fully translated protein, which includes the 19-residue signal peptide, is frequently used in the literature when referring to genomic data and clinical case reports. For example, the fully translated protein numbering for the codominant allelic variant of PLG is written as p.D472N, while the corresponding mature protein number is PLG/D453N (lacks the signal peptide). In this review, we used the mature protein numbering for all PLG variants in the text. To enhance comparison with data from the literature, the fully translated and the mature protein numbering for PLG variants are stated side-by-side in the Tables.
The mature PLG protein (Glu1-PLG) is multi-modular, containing consecutively from the amino terminus (Figure 1): a 77-residue activation peptide (AP), followed by five ∼80 residue triply disulfide-linked kringle (K) domains separated by variable length inter-kringle residues; an activation cleavage site (R561-V562) susceptible to the catalytic cleavage activity of plasminogen activators (PAs), and a light chain homologous to serine proteases, such as trypsin and chymotrypsin. After direct hydrolysis of the R561-V562 peptide bond, as catalyzed by PAs, such as urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), or indirect activation by bacterial activators, e.g., streptokinase (SK) and staphylokinase (Sak), the final protease, plasmin (EC 3.4.21.7), is formed. Plasmin consists of the plasmin/[K78-R561] heavy chain (HC), containing all five kringles, doubly disulfide-linked to the PLG/[V562-N791] light chain, or serine protease (SP) domain, containing the serine protease catalytic triad, His603-Asp646-Ser741 (11–13). After activation, the resulting plasmin lacks the AP, the removal of which is autocatalyzed by plasmin (14). The HC and the LC are latent in the zymogen (PLG). Also provided in Figure 1 are other derivatives of Glu1-PLG, which have occasionally been described in the literature, e.g., mini-PLG and micro-PLG (μPLG), but these are proteolytic products of native PLG, or cloned fragments of this protein, and are not further discussed herein. The post-translational product, Lys78-PLG, is an important activation intermediate of Glu1-PLG and will be referred to in this review.
The kringle domains and their lysine binding sitesOf essential importance to PLG/plasmin function, are the five kringle domains of the PLG-HC, four of which, viz., K1, K2, K4, and K5, bind to lysine with varying affinities. Figure 2A shows the x-ray crystal structure of the binding of a lysine analog, ε-aminocaproic acid (EACA), to isolated PLG-K1 and the figure highlights the critical lysine binding residues (15). Figure 2B represents a generic 79-residue lysine binding kringle, based on the numbering in PLG-K1. The location of the important lysine binding residues for each of the kringle modules of PLG is summarized in Table 1. Of course, other residues can assist in the stabilization of the ligand, but the residues shown are important for binding in each of the kringle domains.

Figure 2 The essential binding residues for a LBS to be present in a kringle. (A) The x-ray crystal structure of the binding of a lysine analog, ε-aminocaproic acid (EACA), to isolated PLG-K1(PDB ID, 1CEA). Asp(D)/Glu(E) side chains at residues 54 and 56 (numbering beginning at Cys1 of the kringle) are positioned to interact with the ε-amino group of EACA and Arg(R)70 bridges the COOH group of EACA. Aromatic residues at amino acids Trp(W)61 and Tyr(Y)71 stabilize the central methylene groups of EACA. Tyr(Y)63 forms a hydrogen bond with the COOH group of EACA. (B) A generic 79-residue kringle (based on PLG-K1) is shown emphasizing the locations of the critical amino acids that are needed for strong binding.

Table 1 Critical amino acids/centers necessary for the lysine binding function of each kringle domain of PLG. EACA is used as a lysine analog.
There are three major centers within the LBS that are essential for the lysine-binding event (Figure 2A). Firstly, an anionic center, formed by two aspartates, Asp54 and Asp56 (numbering beginning at C1 of the generic kringle), that coordinate with the amino group side chain of lysine, lysine isosteres, and lysine analogs, such as EACA. Notably, one of these aspartates is replaced by glutamate in PLG-K2 and by lysine in PLG-K3. Secondly, a hydrophobic core center, in which two aromatic amino acids, in this case, Trp61 and Tyr71, form a cluster that stabilizes the central methylene groups of EACA, and lastly, a cationic center, composed of basic residue(s), which interact with the carboxylate group of the ligand. As shown in Figure 2A, Arg70 interacts with the COOH group of EACA, while Tyr63 not only supports the hydrophobic core but also forms a hydrogen bond with the COOH group of EACA. Multiple studies indicate that residue-to-residue variations among the kringle domains, highlighted in Table 1, affect their lysine binding affinities.
Additionally, at least for the binding of EACA to K1-PLG, an Arg at position 34 further stabilizes the carboxyl group of the ligand. Phe35 contributes to the hydrophobic cluster that surrounds the backbone of EACA, while the side chains of Tyr71 and Tyr73 support the anionic center by having interatomic distances that suggest that it can serve as a hydrogen binding partner for EACA and for Asp56, respectively, thus stabilizing this latter residue in the lysine binding pocket (Figure 2A). In studies with the isolated kringle domains, PLG-K1 has the highest lysine binding affinity, followed by PLG-K4, PLG-K5 and PLG-K2, while PLG-K3 poorly binds to EACA (16), due to the presence of a lysine (Lys56) instead of the acidic amino acid side chain, Asp56 (Figure 2B), in its anionic center. Further, PLG-K5 contains Leu70, rather than Arg70 in its cationic center, a factor that likely governs its weaker binding to EACA (17, 18).
Functions of LBS in receptor binding and regulation of the PLG conformationThe lysine binding sites (LBS) of kringle domains are critical for the functional properties of PLG and allow PLG and plasmin to bind to cellular receptors utilizing C-terminal lysine residues (19) or internal through-space isosteric lysines formed from proper spacing of amino acid side chains (20). This binding activity stimulates the activation to plasmin and places the potent protease, plasmin, on cell surfaces where it is also resistant to inactivation by natural inhibitors, e.g., α2-antiplasmin (α2AP) and α2-macroglobulin (α2M) (21).
The PLG/plasmin system is primarily involved in the degradation of fibrin but also is a key participant in other proteolytic migratory cellular functions, including tissue repair, extracellular matrix degradation, angiogenesis, tumor invasion, inflammatory cell migration, complement protein interactions, and in maintaining healthy body mucosal surfaces by removing fibrin and misfolded proteins from extravascular tissues (22–27). The PLG activation system is tightly regulated by serpin inhibitors of PAs, such as plasminogen activator inhibitors-1 (PAI-1) and -2 (PAI-2).
Since any free plasmin generated in plasma would be rapidly inactivated by circulating protease inhibitors, most of the pathophysiological cell migratory functions of plasmin, e.g., wound healing, employ cell-bound plasmin. Thus, specific PLG/plasmin cellular receptors are needed. In mammalian cells, glycolytic moonlighting proteins, such as enolase, play important roles in this regard (19, 28–32), whereas in microbial cells, surface proteins, such as M-protein, and even enolase, which migrates from the cytoplasm to the cell surface by an unknown mechanism, are important PLG receptors used by bacteria for migration and dissemination (33).
PLG closed (T) and open (R) conformationsNot only do the lysine binding sites (LBS) of kringle domains mediate PLG interactions with other proteins, but also basic amino acid side-chains within the AP interact intramolecularly with LBS' of kringle domains, esp., K2-PLG, K4-PLG, and K5-PLG, to place PLG in a tight (T) poorly activatable conformation (34–36). Biochemical and biophysical studies, in addition to the x-ray crystal structure of PLG (37), indicate that the LBS residues of intact PLG, viz., Asp411 and Asp413 (equivalent to Asp54 and Asp56 of the isolated PLG-K4), make several interactions with Arg68 and Arg70 in the AP domain. Additionally, Asp518 in the anionic center of the PLG-K5 interacts with Lys50 of the AP domain. Likewise, in the LBS of PLG-K2, Asp219, Glu221, and Arg234, interact with other residues located in the SP domain of PLG. These interactions serve to place PLG in a tightly folded and closed activation-resistant T-conformation (32, 38–40), thus maintaining PLG in plasma, which otherwise would be activated, with the resulting plasmin rapidly inactivated by circulating inhibitors. Upon binding to cellular receptors via the LBS, the intramolecular interactions between the LBS' and the AP and SP residues are displaced, inducing a change that relaxes the conformation of the bound PLG (R) rendering it highly activatable (41). This step results in an increased susceptibility of PLG (R) to convert to Lys-PLG by the cleavage of the exposed Arg560-Val561 peptide bond by plasminogen activators. Most recently, systematic inactivation of critical LBS residues in the various kringle domains of PLG was used to determine their effects on PLG activation by tPA, uPA, or SK. The results indicated that the LBS of PLG-K2 has the highest influence on relaxing the PLG conformation and enhancing its activation potential, followed by PLG-K4 and PLG-K5, with PLG-K1 having the smallest influence (32).
PLG post-translational variantsSeveral posttranslational variants of PLG are found in plasma samples and also in purified preparations of the protein. Without inclusion of protease inhibitors in the purification media, a portion of the native Glu1-PLG can be converted to Lys78-PLG by proteolytic removal of the 77-residue N-terminal AP (Figure 1). The product, Lys78-PLG, is far more activatable to Lys78-plasmin than is Glu1-PLG, but both forms of PLG are converted to the same plasmin, viz., Lys78-plasmin (14, 42). Another source of variation in PLG is the two glycoforms separable by specific affinity chromatography on Lysine-Sepharose. This is a general feature of plasminogens from plasmas of different mammalian species (43). These glycoforms have been characterized as a population of PLG not N-glycosylated at Asn289 and another form of PLG which is glycosylated with N-linked biantennary complex carbohydrate at Asn289. Thr346 is fully O-glycosylated in the entire PLG population (7, 8, 44). The properties of these glycoforms have been studied extensively since their discovery and differences between them have been found in lysine binding, PLG activation rates, fibrin binding, and catabolic rates (43, 45–47). In addition, isoelectric focusing (IEF) reveals a number of PLG subspecies ranging in pI from 6.4–8.5 that are primarily derived from differences in sialic acid content on the carbohydrate. Treatment with neuraminidase reduces the number of these bands (45, 48).
Since these variations of PLG are not allelic variants, but are post-translational modification subforms, they will not be further considered in this review.
PLG polymorphismsMutations found in the PLG gene include nonsense, missense, frameshift, splice site, deletion and insertion variants that can affect the structure and function of the PLG protein zymogen and its activated product, plasmin. In this review, we focus on missense variants and, to the extent possible, discuss the mechanisms by which these variants can affect the structure and function of PLG and plasmin.
PLG contains several relatively abundant alleles with non-synonymous single nucleotide polymorphisms (nsSNP) that result in missense variants. Some of these alleles appear globally, while others are restricted to different populations. Most of the common PLG missense variants are not thought of being directly deleterious. However, they may contribute via a cumulative effect to increase disease risk when in combination with other PLG variants, or with other protein pathogenic variants and with environmental factors (49, 50). Because PLG plays a critical role in inflammation and disease, it is important to be aware of major PLG variants in the population and their potential effects of PLG/plasmin dysfunction.
The fibrinolytic potential and plasmin generation capacity in individuals can vary significantly and this fact requires attention as to which fibrinolytic drugs should be used in different patients (27). An earlier study reported that the ability to generate plasmin can vary 8-fold in healthy individuals in addition to differences attributed to gender, age, and the use of contraceptives (51). It is not clear whether the existence of polymorphic PLG contributes to some of this variation. The ability to activate PLG to plasmin using different PAs needs to be considered before administering therapeutic treatments to patients carrying certain PLG variants. Understanding the relative world abundance and potential phenotypical consequences of relevant PLG variants is therefore of interest to medicine and population biology, as well as forensics.
Minor allele frequency (MAF)In population genetics, the most common allele for a given SNP is referred to as the major allele, while less common alleles are termed minor alleles. The frequency of occurrence of the less common allele (aka, the second-most common allele) is presented as the Minor Allele Frequency (MAF). The MAFs are useful as they provide information about how common a particular SNP is within a given population. The MAF often varies geographically, and both global and regional numbers are important and useful when focusing on populations or resulting protein variants encoded by the allele. Rare alleles are prone to appear locally while common alleles are shared over a wider population range (52).
In this review, MAFs are classified into four groups based on relative abundance ranges:
(1) Polymorphisms: those variants with MAF% ≥5%, corresponding to a MAF ≥0.05.
(2) Common variants with MAF% = 1%–5%, corresponding to a MAF of 0.01–0.05.
(3) Low frequency variants with MAF% = 0.1%–1%, corresponding to a MAF of 0.001–0.01.
(4) Rare and ultra-rare variants with MAF% ≤0.1%, corresponding to a MAF ≤0.001.
While many rare Mendelian diseases are caused by rare (and ultra-rare) variants with large effects, it is believed that both rare and common variants with smaller effects play roles in both complex diseases, but how they work together is unclear (49). Low frequency variants have an important impact in the phenotypic variation at a population scale (53). Genome-wide association studies (GWAS) cannot fully explain the heritability of complex traits (54). This missing heritability effect can be explained by common variants having a weak effect in combination with low-frequency rare variants, which together can lead to complex diseases (55).
PLG polymorphisms: historical contextThe term PLG polymorphisms was first used in the 1970s when researchers started to discover PLG protein variants. During this period, there was no information about MAFs, and PLG polymorphisms were only defined by PLG protein variants carried by the population. Moreover, phenotyping of PLG became of great interest when an abnormal PLG with an unusual electrophoretic mobility pattern was reported in a patient with recurrent thrombosis (56).
Other PLG polymorphisms have since been described using isoelectric focusing (IEF) gel electrophoresis (57, 58). Usually, the procedure to detect PLG polymorphisms by IEF involves treatment of patient plasmas with neuraminidase to remove negatively charged sialic acid from glycan structures and reduce the complexity of the isoforms. The treated plasma is usually next submitted for IEF gel electrophoresis at a pH range 3–10 (or 5–8). PLG is then functionally assayed by activation with uPA or SK with a chromogenic substrate-containing assay kit, and/or by following the lysis of casein in an agar overlay (59). PLG patterns are often obtained by immuno-detection and Western blots.
The interest in PLG polymorphisms increased upon observations that ethnically different populations presented with dissimilar frequencies for certain PLG variants as detected by IEF (60). Accounts of PLG variants in individuals started to accumulate mostly between 1970 and 2000 (56, 61, 62). Some polymorphisms were initially confirmed by amino acid sequencing (63). The PLG/D453N polymorphism was identified when the PLG gene was first characterized (5). The phenotypic distribution for the PLG/Asp453 and PLG/Asn453 alleles was found to fit the Hardy-Weinberg equilibrium, with an autosomal codominant inheritance matching a Mendelian inheritance mode (64).
To identify the many different PLG phenotypes discovered from individual plasmas, an alpha numeric nomenclature system was proposed (65). This nomenclature is based on using as a reference the IEF mobility of the two most common PLG polymorphic codominant alleles. They were initially labeled, PLGA, with A for acidic, and PLGB, with B for basic. Other alleles were compared to A and B mobilities in terms of being more acidic or more basic than these major forms. The identification therefore included A-like and B-like designations. The letter M was used to refer to an intermediate variant or medium (between A and B) and C was used for common (65). It was soon realized that PLG-based allelic signatures could help generally identify an individual. This gave rise to the use of PLG polymorphisms in forensic hemogenetics, which included paternity examinations (59, 66, 67). It was later found that the polymorphic IEF phenotypes, PLGA and PLGB, were generated by a single amino acid substitution of the more acidic PLG/Asp453 for the relatively more basic PLG/Asn453, respectively (68). These polymorphisms were included in many PLG deficiency (PD) case reports and became a reference for the IEF phenotype nomenclature (68).
Prior to standardizing this nomenclature, PLG polymorphisms were difficult to refer to and to compare. Different designations were initially given, including the city of origin of the patient. For example, PLG-Tochigi (69), a mutant with reduced plasmin activity after normal activation, was identified as IEF-M5 and later associated to PLG/A601T. PLG-Osaka also produced a PLG variant that led to a form of plasmin with reduced activity (70). This was classified as IEF-M and later identified as PLG/D676N. The most frequent IEF patterns often included combinations of one or two wild-type (WT) PLG alleles with a combination of one or two common PLG alleles. Overall, about eighteen phenotypic PLG polymorphisms were initially identified using IEF (71). Other names for variants included PLG-Nagoya (72), PLG-Chicago (73), PLG-Frankfurt (74), and Plasminogen Paris (75). On occasion, phenotypes were identified with designations such as PLG-1 which was later associated with the A-phenotype. Case reports of novel PLG polymorphisms after the year 2000 occasionally use the city of origin of the proband. The PLG-Kanagawa-I polymorphism was reported in 2002 and corresponds to a dysfunctional PLG activity caused by the PLG/G732R variant (76).
PLG phenotyping based on the IEF protocol has several advantages, viz.: (1) PLG is readily available from patient plasmas for further characterization; (2) the PLG protein band pattern corresponding to the translated alleles from the blood of an individual can be readily visualized; (3) the electrophoretic mobility provides information about the overall charge of the protein as compared to wild-type (WT)-PLG and differences can be an indication of amino acid changes and different alleles; (4) many times an allele is expressed in a lower amount and the relative abundance of alleles could provide phenotypic information; and (5) the isoelectric point for a protein with a known amino acid sequence can be calculated. Since IEF changes may reveal alterations of the PLG structure, the IEF pattern adds valuable information for a phenotypic characterization of a PLG variant in a patient and a first step towards a diagnosis of a PLG deficiency.
The IEF protocol helped to visualize the existence of various PLG phenotypes in plasma and PLG variants sometimes associated with disease. PLG genetic analysis was later introduced, especially when young individuals presented unusual symptoms, e.g., thrombosis, which made the search for abnormalities in this gene a valuable approach. The need to purify PLG variants for further analysis was also suggested when subjects were found to carry different IEF patterns (77).
Whereas IEF analysis is still often used as a characterization step, this is usually followed by genomic DNA analysis of PLG, including the use of the polymerase chain reaction (PCR), single-strand conformation polymorphism (SSCP) analysis, and/or direct DNA sequencing (68, 78). A summary of several IEF phenotypes with corresponding PLG molecular variations has been reported (79).
IEF from case reports of patients and families, combined with DNA sequence information, has contributed to the discovery of many amino acid substitutions in PLG deficiencies. IEF, followed by DNA sequence analysis, was most recently used in the discovery of the PLG/K311E missense variant that leads to a rare disease known as hereditary angioedema (HAE) with normal C1 inhibitor. This variants has been cataloged as a clinical variant (80).
Predictive algorithms of protein dysfunctionMost PLG missense variants of interest lack functional studies and their clinical significance are missing or uncertain. Amino acid variants can range from benign to pathogenic. Predictive in silico computational methods can provide highly likely scenarios of amino acid substitutions in proteins, especially when using different approaches (81–83). To facilitate a more comprehensive discussion of the pathogenic variants that will be discussed later in this review, we consider it essential to conduct an in silico analysis that predicts potential structural and functional perturbations resulting from various amino acid substitutions in PLG variants. This analysis will enable us to better understand the molecular implications of these variants and provide valuable insights into their pathogenic potential. Herein, we used the following in silico prediction tools. SIFT (Sorting Intolerant From Tolerant), which is based on sequence conservation (84); Polyphen-2, which assesses the impact of amino acid substitutions on protein structure/function (85); mCSM, which predicts the effect of variants in proteins using graph-based signatures (86); MUpro, which predicts protein stability changes based on protein sequence and structure and uses Support Vector Machine (SVM) (87); and DynaMut2, which combines Normal Mode Analysis (NMA) methods to capture protein motion and graph-based signatures (88). PLG structural data used for mCSM, MUpro, and DynaMut2 was based on the x-ray structure of Glu1-PLG (PDB ID, 4DUR) (36) and the cryo-EM structure of PLG (PDB ID, 8UQ6).
For the amino acid substitution effects using Polyphen-2 and SIFT, the score for substitution of each residue in each prediction tool was first recorded. A red-green heat map was then created from those values by assigning bright red for the most damaging score and bright green for the most tolerated score for each prediction tool. Specifically, Polyphen-2 score 0, benign (green); score 0.5 (mid-range), possibly damaging; score 1, probably damaging (red). SIFT score < 0.05 is predicted to be deleterious (red); score 0, variants can affect protein function (red); and score 1, tolerated (green). We excluded nonsense variants since stop codons cannot be modelled with the prediction software. Clinical variant classifications were based on the ClinVar (NHLBI) algorithm (https://www.ncbi.nlm.nih.gov/clinvar/). Accession numbers for PLG missense clinical variants are provided in the text as appropriate.
The high-resolution structures of PLG have contributed greatly to the understanding of its structure/function relationships and facilitates making credible functional predictions. Studying the effects of single amino acid substitutions in PLG that lead to clinical outcomes, as found in congenital PLG deficiencies, also presents a convenient informational source that can provide critical insights into its role in vivo. Animal models, such as PLG gene-altered mice (89, 90), in combination with various other related transgenic murine models, continue to be instrumental in understanding the mechanisms of PLG function.
Prevalence of PLG missense variants in different populationsIn general, population data of a variant is important when evaluating its pathogenicity. Usually, the most abundant variants are not directly pathogenic but may contribute in a minor way to complex diseases, especially if the variant is predicted as pathogenic and if it occurs in a protein like PLG which is involved in many disease mechanisms (50). The chances of a pathogenic condition increase in homozygous or compound heterozygous states where the additive effect increases the penetrance (50).
The gnomAD browser v4.0.0 currently lists ∼1,000 missense PLG variants detected from a wide variety of large-scale sequencing projects (https://gnomad.broadinstitute.org/). Most of the PLG nsSNPs are rare or ultra-rare (MAF≤ 0.1%), while less than 2% of the variants (Table 2) are relatively abundant (MAF ≥0.1%) in various genetic ancestries in the world. From the 2% group, most major PLG missense variants are assumed to be benign, but in fact, not much is known about them at the molecular level and, therefore, are also of great interest in this review.
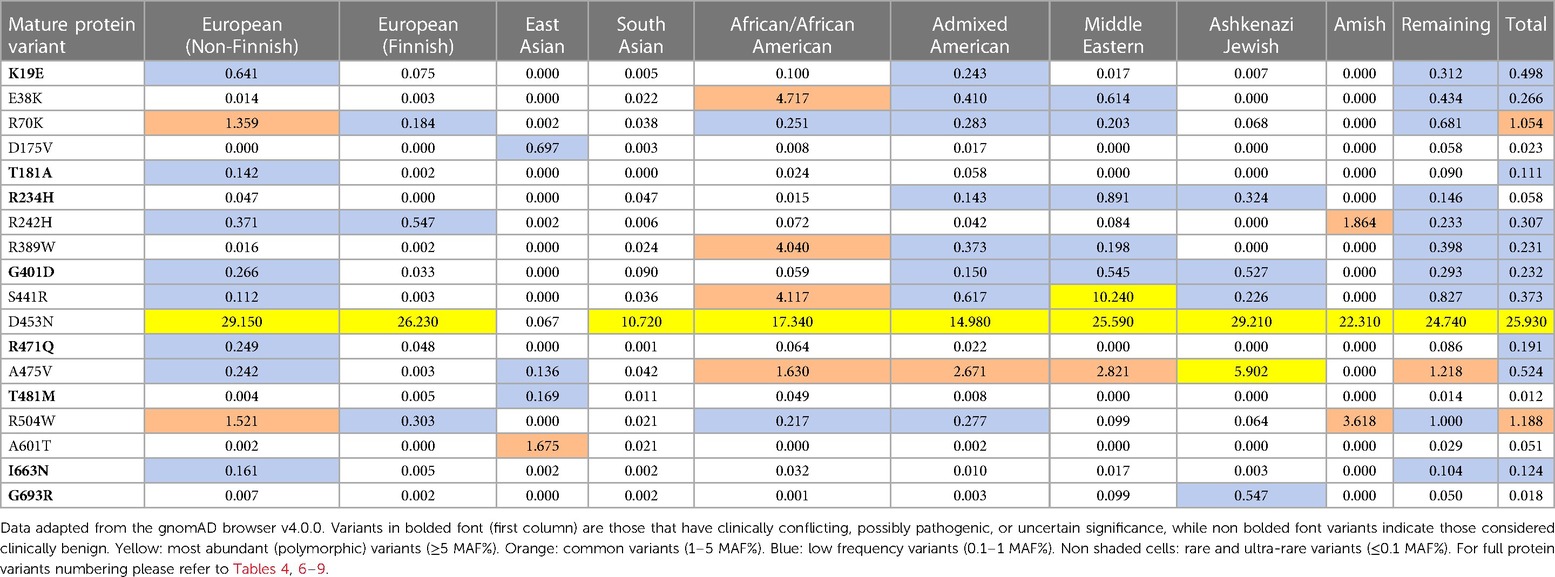
Table 2 MAF percent distribution of major PLG missense variants per genetic ancestral group (gnomAD database).
In addition to the data from gnomAD browser v4.0.0, the data obtained from the PAGE population study (Table 3) are included in this review because they provide access to genomic data from various American populations involving various races and ethnicities that have not been sufficiently represented in a world in which diversity is progressively increasing (91). The multiscale nature of the MAF% distribution of major PLG variants in different ethnic backgrounds is evident in both Tables 2, 3. The PAGE population study compiles allelic data from various populations, including Native Hawaiians and Native Americans, not readily available in the past (91), and studies on a genetic propensity for stroke in such populations can now benefit from these data. As an example, it has been recently reported that these populations have a higher-than-normal propensity to stroke at younger ages with significantly higher stroke mortality in comparison to other regional ancestries in local populations (92).
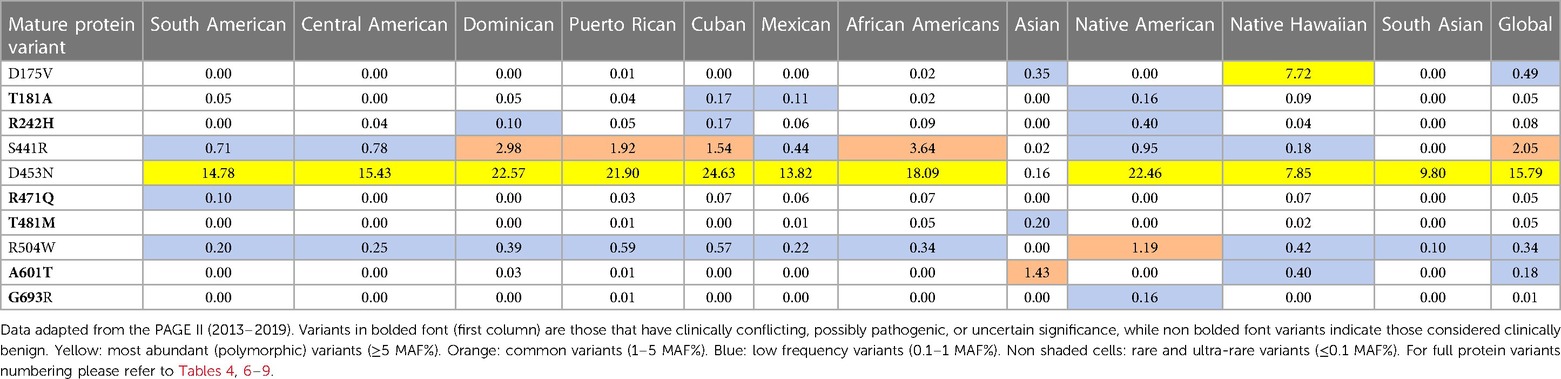
Table 3 MAF percent distribution of major PLG missense variants per genetic ancestry group (PAGE database).
The overall relative distribution trend found with the gnomAD browser for the second minor allele for PLG missense variants was consistent with two other populations studies, the ALFA project (https://ncbiinsights.ncbi.nlm.nih.gov/2020/03/26/alfa/), and with the 1000 Genomes project (https://www.ncbi.nlm.nih.gov/bioproject/28889).
All the PLG missense variants discussed in the present review are illustrated in Figure 3 showing their location along the primary structure of the PLG protein.
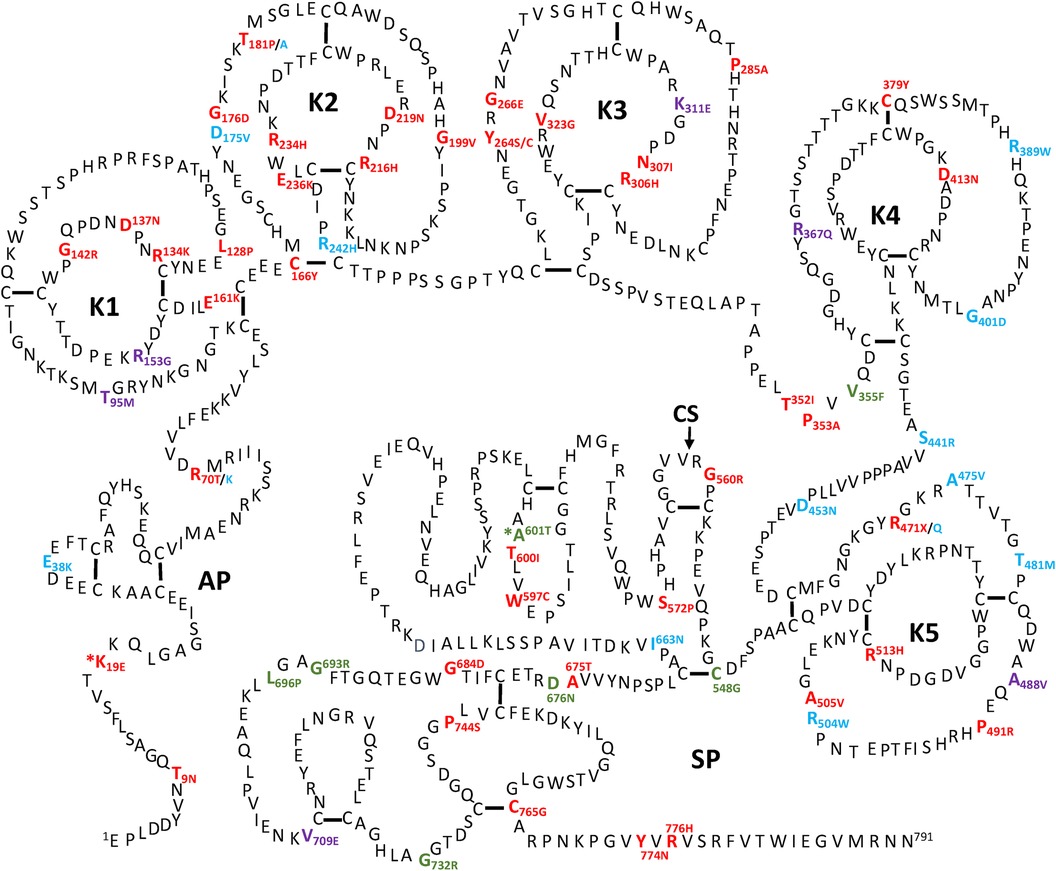
Figure 3 Placement of the known PLG variants within the PLG primary structure. Missense variants are designated red in PDI and green in PDII (green). Major PLG missense variants in the population are presented in blue. Rare pathogenic/or possibly pathogenic PLG missense variants associated with other various disorders (purple). The asterisk in *K19E (PDI) and *A601T(PDII) indicates that these two variants are also relatively abundant. For full protein numbering of PLG variants refer to the Tables 4, 6–9.
Missense variants and plasminogen deficiency (PD)PD-associated PLG missense variants have attracted much attention, and they are among the most described in the literature. Except for the relatively abundant PLG/K19E and PLG/A601T variants, most PD-associated nsSNPs are rare or ultra-rare and are not necessarily always detected in population studies. In fact, most are described in case reports from diseases running within families.
Two types of PD, which include type I (PDI) and type II (PDII), have been described. The PDI and PDII missense variants are represented in red and green font, respectively, in Figure 3. Notably, PLG/K19E and PLG/A601T are both abundant and related to PD and those have an added asterisk to represent this duality. Single gene Mendelian diseases like PD offer a unique opportunity to study protein structure/function in relationship to disease phenotype.
PLG deficiency-type I (PDI)PDI, also known as true PLG deficiency or hypoplasminogenemia, is a genetic disease characterized by low or undetectable PLG antigen. Thus, reduced plasmin activity in plasma is found. This condition results in compromised fibrin clearance (93). Congenital PDI is mostly inherited as an autosomal recessive trait and it is cataloged as a rare disease by the National Organization of Rare Disorders (NORD) (https://rarediseases.org/).
To date, about 45 single amino acid substitutions in PLG have been discovered in probands with PDI. Table 4 summarizes the amino acid substitutions reported for PDI, their domain mapping, and the result of our in silico predictions. The current clinical significance for the majority of these substitutions is either missing or has conflicting interpretations.
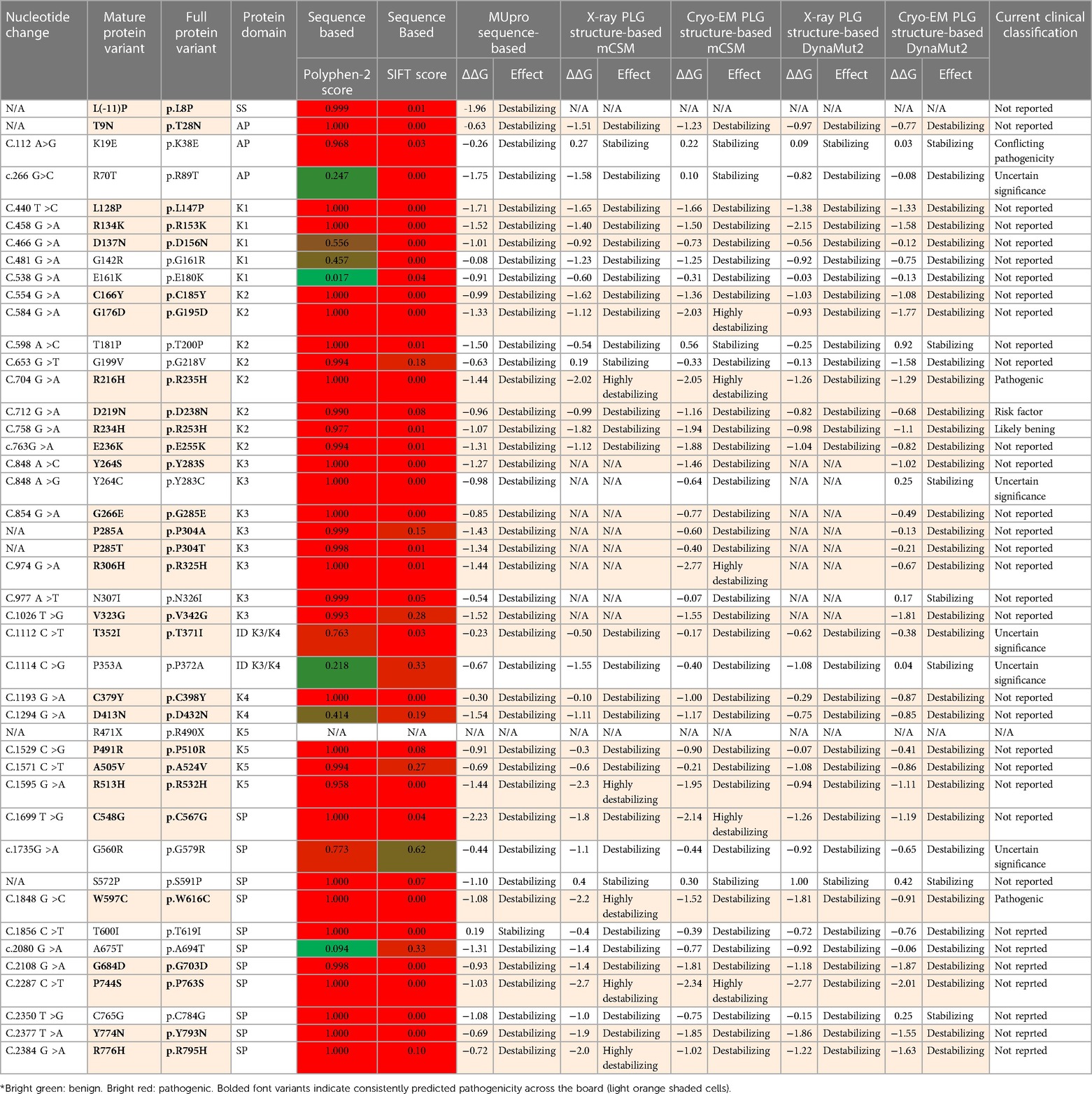
Table 4 PLG missense variants reported in association with PDI with domain locations and predicted sequence-based and structure-based amino acid substitution effects*.
Clinical manifestations of PDIThe reduced PLG antigen concentration and/or activity characteristic of PDI leads to extravascular accumulation of undigested fibrin and impairs wound healing in mucosal surfaces. This debris causes thick white-yellowish pseudomembranous lesions with a wood-like (ligneous) appearance. Histologically, pseudo-membranes show accumulation of hyaline-like substances, impaired epithelial and fibrin debris with inflammatory cells, fibroblasts, and eosinophilic infiltration (25). The dominant presentation of PDI occurs in the eye-lid surface or conjunctiva, known as ligneous conjunctivitis (LigC), which accounts for up to 80% of the clinical presentations. Similar systemic lesions may occur in additional mucosal tissues, including the gingiva (known as ligneous gingivitis or LigG), the middle ear, the larynx, and the female genital tract. Periodontal disease can be the first clinical manifestation of LigC and PDI (94). Approximately 60% of cases of LigC also develop LigG in PDI with∼a 2:1 ratio of female to male presentation (95). The PLG/K19E mutant has been reported in 34% of the LigG cases (95).
Table 5 (Cases A to D) summarizes data from four previously reported case studies involving PLG nsSNPs associated with PDI. The data were adapted from case reports and reviews of patients and family members with PDI with no other known health conditions. Notably, data from case reports are often missing critical information. Where available, %PLG activity, %PLG antigen, zygosity, gender, reporting age, and phenotypes are presented. From these available data, PDI symptoms become evident when the %PLG activity is <40%, regardless of the variant. Also, in most cases, homozygotes or compound heterozygotes for certain PLG nsSNPs were needed for clinical manifestations of PDI. This is true for PLG/K19E, PLG/R216H, PLG/W597C, and PLG/L128P, suggesting an additive effect and different penetrance. PLG/K19E may have lower penetrance since at least one homozygous relative did not show clinical manifestations. Variable penetrance and expressivity of variants is a recognized limitation that affects our understanding of the effect of a variant when comparing case reports with the population at large (99). Occasionally, a variant may be sufficiently pathogenic in a given individual to be able to cause clinical symptoms in a heterozygous state, as is the case for PLG/A505V variant in Case A, patient 2, in Table 5.
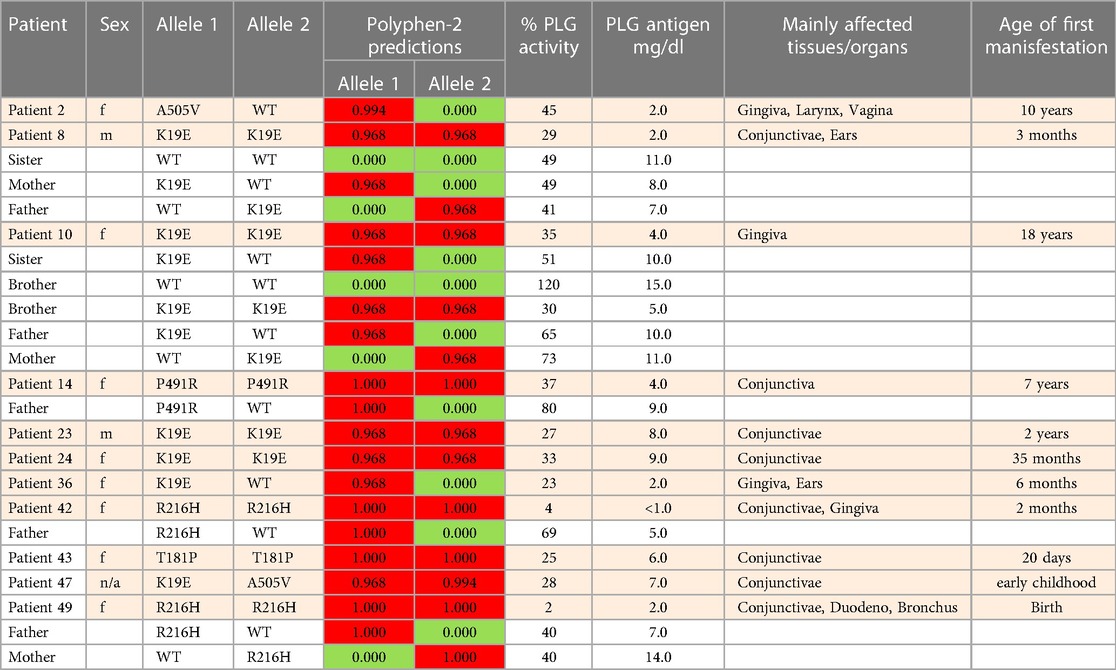
Table 5A Clinical manifestations of PDI missense variants and prediction analysis for several case studies.
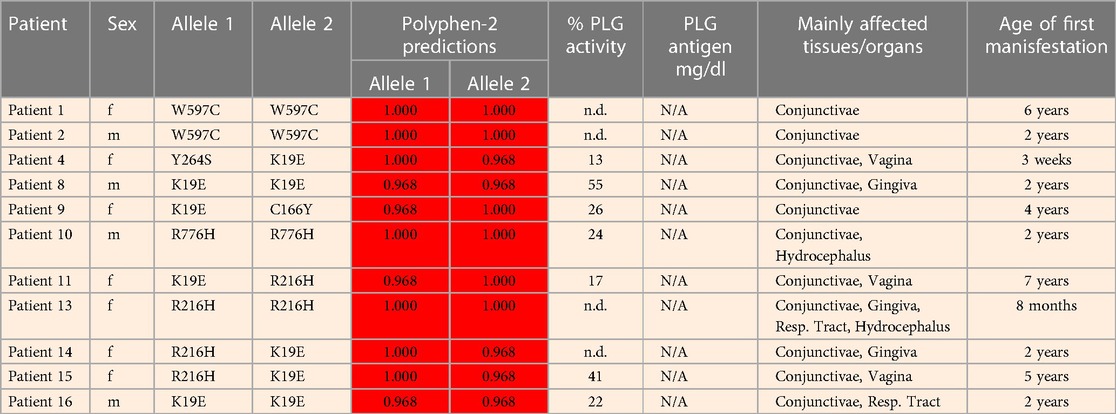
Table 5B
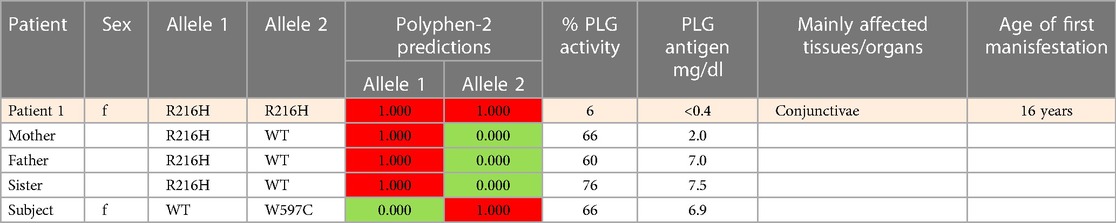
Table 5C
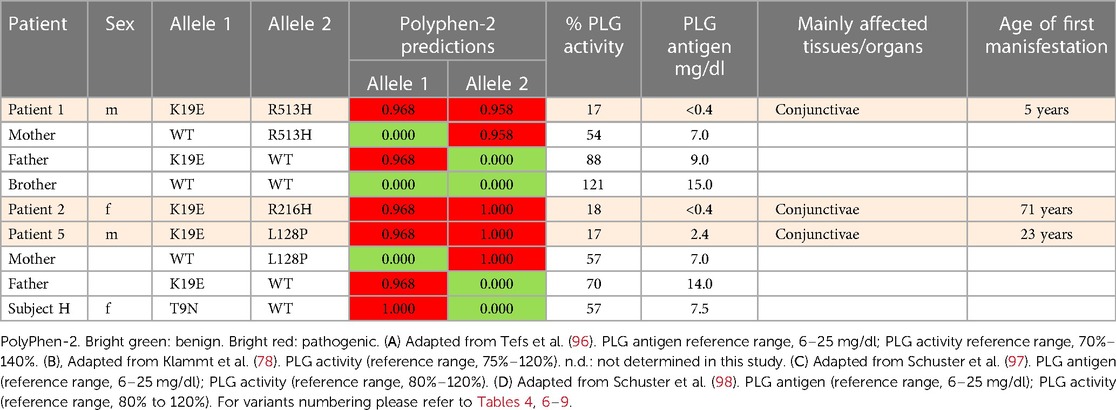
Table 5D
An important finding resulting from case studies of heterozygous relatives of congenital PLG deficiency patients is that PLG activity and PLG antigen concentration can be significantly lower than that considered to be normal, with individuals appearing healthy. As an example, the PLG antigen concentration could go as low as 2 mg/dL, with no disease phenotype reported, when having 66% PLG activity (Table 5, Case C, Patient 1, mother). An estimate of how low the PLG activity can be without disease phenotypes based on these cases is about 50%. When the PLG activity and antigen concentration of PLG are too low, clinical manifestations of quantitative PLG deficiency become evident. Beyond a threshold level, symptomatic patients present extravascular fibrin deposition and systemic inflammation.
Overall, PDI can result in severe consequences, including blindness, tooth loss, and infertility in both males and females (25, 100). This debilitating illness significantly impacts quality of life (101). Furthermore, as a rare disease, PDI poses diagnostic challenges worldwide, resulting in delayed access to limited but potentially life-changing interventions (102).
Prevalence of PDIThe overall prevalence of congenital PD as homozygous or compound heterozygous is reported to be 1/625,000 (orphanet.net). These numbers are expected to be high within regions where consanguineous unions are common. Notably, about 0.13%–0.42% of the world population can be asymptomatic heterozygous carriers of PD alleles (103). This is an important issue since most PDI-associated variants are predicted to be pathogenic and, as such, they may contribute to disease even in heterozygous individuals. Such pathogenic variants may add to the variation in PLG levels and activities observed in the population and possibly contribute to complex, non-Mendelian diseases.
A significant number of patients with PDI are of Turkish origin (25, 93, 104). Turkey has a 19% consanguineous union frequency, with 58% of those being between first-cousins (105). One report showed that 21 of 50 studied patients were of Turkish origin with consanguineous union between parents in 19 members of that study group (96). The Middle East also presents some of the highest rates of consanguinity in the world, with Arabian first cousin consorts reaching 25%–30% of all marriages (106). Likewise, there is a high rate of common ancestral unions in inner Asia (107) and North African countries (108). Unfortunately, allele databases from these regions are not readily available. There are ongoing efforts to improve the current limited access to genomic data from Lebanon and Africa (109, 110). India is also known to have a high burden of rare recessive genetic diseases (111). Importantly, the most recent version of the gnomAD browser (v4.0.0) now includes Middle Eastern ancestry data, which is known for having high consanguinity.
Potential mechanisms of PDIIt is not clear how the different PLG missense variants lead to PD and the nature of the molecular mechanisms are equally uncertain. Most of the PLG variants associated with PDI have not been fully characterized beyond IEF. Herein, we utilized prediction software to assess the impact of the resulting substitutions on PLG structure and function. It is believed that the structural changes that occur in PDI variants may result in an impaired secretion and/or a reduction in half-life. Unfortunately, the half-lives of most known PDI variants have not been reported.
As indicated by the red-green heat map in Table 4, most PDI substitutions were predicted to be damaging (bright red) for the sequence-based predictions Polyphen-2 and SIFT. The ΔΔG values from the structure-based predictions are consistent with most PDI substitutions being destabilizing when using either protein data base (PDB) structure files. It is seen from this Table that the majority of the PDI variants are located in the very conserved kringle domains of PLG (Column 4, Table 4 and Figures 4A,B) which would destabilize the protein. Interestingly, several PDI variants were consistently predicted to be highly destabilizing. Those variants include PLG/R216H in K2-PLG, PLG/R513H in K5-PLG, and three variants (PLG/W597C, PLG/P744S, and PLG/R776H) in the SP domain of PLG/plasmin (Table 4). These predictions are consistent with recent findings that show that a functional LBS in K2-PLG is critical to maintain the PLG closed conformation by interactions with the SP domain, and that the LBS’ in K4-PLG and K5-PLG are also critical to maintaining the activation resistant form (32). PDI variants in the SP domain probably mostly destabilize the closed conformation leading to PLG short half-lives. About 20% of PDI substitutions occur in the SP domain (Figure 4B). Herein, we discuss the potential pathogenic mechanisms of some of the variants.
留言 (0)