
記住我






tRNAs play an essential role in translation, mediating the conversion of nucleic acid templates into protein products. They facilitate this by binding a programmed amino acid substrate before transferring it to the ribosome for incorporation into a peptide. During this process, tRNAs make specific interactions with several binding partners: elongation factors, mRNA, and multiple sites within the ribosome. As such, tRNAs directly influence translation and are a prime scaffold for its engineering. This engineering primarily addresses three fundamental applications: i) basic research, ii) genetic code expansion (GCE) in synthetic biology, and iii) tRNA therapeutics.
Basic research on understanding protein synthesis has identified a plethora of information regarding tRNA sequence and structure. The specific elements in the tRNA sequence that promote or deter interaction with a binding partner are termed identity or anti-determinant elements, respectively. The interactions between tRNAs and their cognate aminoacyl-tRNA synthetase (aaRS) in particular have been extensively studied (Giegé and Eriani, 2023) and are an important first step in translation. Still, aminoacyl-tRNAs (aa-tRNAs) must be efficiently incorporated into the ribosome to complete translation. Natural translation machinery is flexible enough to efficiently incorporate the natural amino acids which contain a broad range of chemical properties. However, ribosomal incorporation of non-canonical amino acids (ncAAs), important for widespread applications in synthetic biology, drug discovery, and basic research, are inherently poor ribosomal substrates that can dramatically lower ribosomal translation efficiency. Chemical synthesis via non-ribosomal peptide synthetases (Walsh et al., 2013; Süssmuth and Mainz, 2017) and post-translational modification enzymes (McIntosh et al., 2009) are some alternate strategies to bypass ribosomal synthesis of these amino acid derivatives. However, these strategies are not compatible with screening vast libraries for structurally unique or medically relevant peptides, which is a possibility in a ribosome-based (Yamagishi et al., 2011; Goto and Suga, 2021; Katoh and Suga, 2022a). Through tRNA engineering, hundreds of traditionally ribosome-incompatible substrates have been permitted for ribosomal installation with varying levels of success (Katoh and Suga, 2022a; Sigal et al., 2024). Additionally, nearly 10% and 50% of pathogenic genetic conditions result from aberrant stop codons (Mort et al., 2008) and missense mutations (Katsonis et al., 2014; Stenson et al., 2017), respectively. This has led to substantial research aimed towards developing modified tRNAs for efficient, orthogonal, and safe readthrough of improper gene sequences (Dolgin, 2022; Anastassiadis and Köhrer, 2023; Coller and Ignatova, 2024).
The process of tRNA engineering itself can be broadly divided into engineering for i) improving the orthogonality of an aaRS:tRNA pair, and ii) moderating the aa-tRNA’s journey to and through the ribosome. Briefly, the former objective optimizes aaRS:tRNA interactions to ensure orthogonality and production of the desired aa-tRNAs (Soye et al., 2015; Tamaki et al., 2018; Melnikov and Söll, 2019; Krahn et al., 2020b; Ganesh and Maerkl, 2022; Kim et al., 2024). The latter objective optimizes aa-tRNA interactions with elongation factors, mRNA, and the ribosome to mediate initiation (Tharp et al., 2021b; Katoh and Suga, 2023a; Katoh and Suga, 2023c) or elongation fidelity (Uhlenbeck and Schrader, 2018; Sigal et al., 2024). Our review addresses tRNA engineering principles for the latter processes as a roadmap for modulating tRNA incorporation into the ribosome. We will discuss four major roadblocks involved in this endeavor: i) initial acceptance of an aa-tRNA into the ribosome (P-site for initiation, A-site for elongation), ii) proper codon-anticodon interaction, iii) peptide bond formation and peptidyl transfer efficiency, and iv) translocation of the tRNA. Through this discussion we will highlight the tRNA elements found to overcome these challenges.
1.1 tRNA structureA tRNA’s overall three-dimensional “L-shape” and specific nucleotide sequence dictates its interactions with binding partners and overall function. Canonical tRNAs vary between 76 and 100 nucleotides (Krahn et al., 2020a) and have a highly conserved “cloverleaf” secondary structure (Holley et al., 1965; Bolton and Kearns, 1975; Berg and Brandl, 2021) divided into five stem-loop arms based on internal base pairing: the acceptor stem, D-arm, anticodon arm, variable arm, and T-arm (Figure 1) (Giegé et al., 2012). The acceptor stem contains the 3′ conserved CCA sequence for attachment of the amino acid and discriminator base that participates in conferring aaRS specificity. The D-arm is named after a conserved, modified dihydrouridine base that aids in stabilizing the tertiary tRNA structure (Maglott et al., 1999). The anticodon arm contains a three-base anticodon sequence for decoding the mRNA surrounded by other nucleotides that impact its efficiency and binding to aaRSs. The T-arm is named after its universally conserved thymine-pseudouridine-cytidine sequence (TΨC) that aids in ribosome and elongation factor interactions (Holley et al., 1965; Nissen et al., 1996). Finally, the variable arm varies in length and function, participating in binding of certain translation factors and/or impacting overall tRNA flexibility during mRNA translation (Sun and Caetano-Anollés, 2009; Krahn et al., 2020a; Prabhakar et al., 2022).
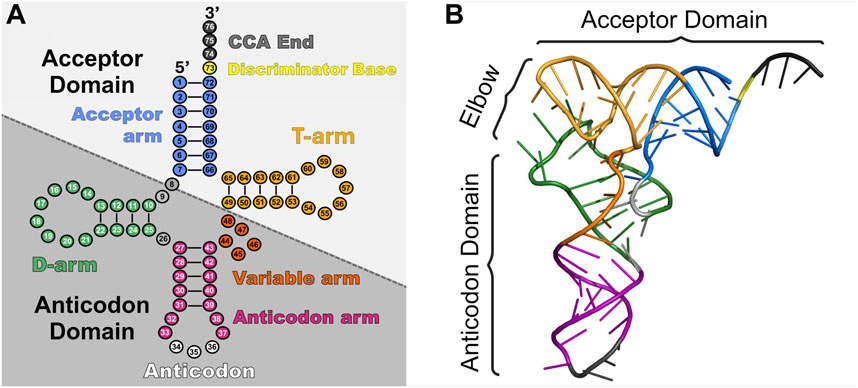
Figure 1. Schematic of tRNA structure. tRNAs consist of two major domains (acceptor domain and anticodon domain) and five stem-loop arms (acceptor arm (blue), D-arm (green), anticodon arm (pink and white), variable arm (orange), and T-arm (gold)) which play different roles in translation. (A) Cloverleaf 2D structure of tRNAAsp (PDB: 6UGG) highlighting the major domains and regions. (B) 3D stick representation of tRNAAsp with domains highlighted.
Structure conservation in tRNAs is also found in 3D (Figure 1), as they twist and fold into an L-shape (Kim et al., 1974; Giegé et al., 2012). This occurs by coaxial stacking of the acceptor stem and T-stem with the D-stem and anticodon stem. Furthermore, the variable loop and D-stem undergo intramolecular tertiary interactions with the D-loop and T-loop (Biela et al., 2023). This newfound L-shape consists of the acceptor domain (acceptor arm and T-arm), elbow region (D-loop and T-loop) (Zhang and Ferré-D’Amaré, 2016), and anticodon domain (D-stem, variable loop, and anticodon arm). The acceptor domain is primarily responsible for binding to its specific aaRS followed by the elongation factor (EF-Tu in prokaryotes, eEF1A in eukaryotes) for transport to the ribosome. The elbow region notably interacts with all three of the ribosome’s tRNA binding domains and an extensive list of RNA and proteins that help mature and modify tRNAs (Zhang and Ferré-D’Amaré, 2016). Decoding of the mRNA occurs through the anticodon domain (Yarus et al., 1986).
In addition to conserved structural features, tRNAs are also heavily modified to further define their function, including all three stages of translation (Agris, 2008; Grosjean et al., 2010; Phizicky and Alfonzo, 2010; Phizicky and Hopper, 2010; Wang and Lin, 2023). In fact, tRNAs are the most extensively modified cellular RNA with about 12% of nucleotides modified (an average of eight modifications per bacterial tRNA and 13 per eukaryotic (Zhang et al., 2022) and around 100 distinct modifications identified in total (Machnicka et al., 2014; Cappannini et al., 2024). Some bases like the 3′ CCA end and discriminator base are never modified, while others are modification hotspots, such as nucleotides in the elbow region (Yared et al., 2024) and anticodon loop (Lorenz et al., 2017).
Our understanding of natural tRNA structure and its impact on translation is imperative to the design of an effective synthetic tRNA. This information lays the foundation for engineering strategies that have been implemented for this purpose.
1.2 Translation processOur focus in translation is tRNA-based, guided in part by structural biology (Chua et al., 2022) which has elucidated many prokaryotic (Rodnina, 2018; Korostelev, 2022; Xu et al., 2022) and eukaryotic translational processes (Neelagandan et al., 2020; Blanchet and Ranjan, 2022; Zhang et al., 2023; Brito Querido et al., 2024). Studies have preferentially investigated the former, resulting in several tRNA engineering strategies developed for prokaryotes. We will focus on the processes of initiation (Figure 2A) and elongation (Figure 2B) as they are most relevant to tRNA engineering efforts.
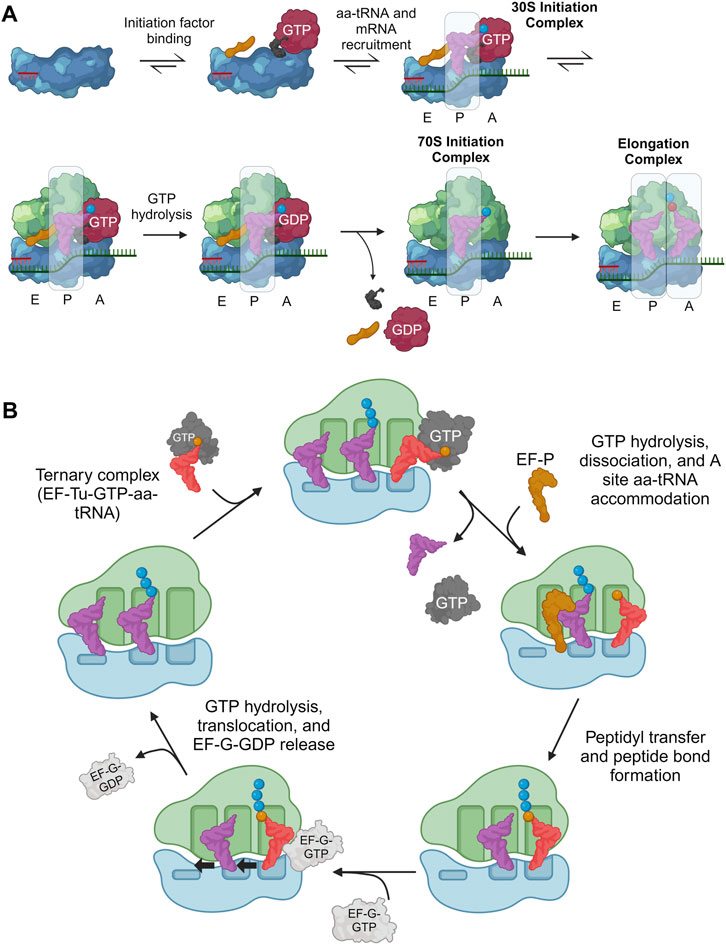
Figure 2. Overview of translation initiation and elongation highlighting important tRNA interactions. During translation, tRNA frequently interacts with both the ribosome and a host of translation factors, playing a central role in all major translational processes. (A) Initiation factors (gold, black, and red) help guide the initiator aa-tRNA into the ribosomal P-site to form the 30S initiation complex. Then, the initiation factors release and allow another aa-tRNA to accommodate into the A-site which catalyzes the first peptide bond to form, marking the beginning of elongation. (B) In translation elongation, the aa-tRNA must bind EF-Tu/eEF1A to be transported to the ribosomal A/T site. Codon-anticodon interactions between the aa-tRNA, mRNA, and ribosome ensure the correct aa-tRNA is selected before peptidyl transfer between the P-site and A-site tRNAs occur. Additionally, the P-site tRNA binds EF-P/eIF5A when tRNAs are charged with difficult ribosomal substrates to improve peptidyl transfer efficiency. Finally, EF-G/eEF2 interacts with the ribosome and A-site aa-tRNA to catalyze translocation. Created with BioRender.com.
Initiation of protein synthesis involves anchoring the ribosome onto an mRNA start codon (often AUG) and inserting the first aa-tRNA into the P-site. In bacteria, initiation is composed of two major processes (Milón and Rodnina, 2012; Roy et al., 2017): i) assembly of the 30S initiation complex (30SIC) which involves the binding of initiation factors (IF1, IF2, and IF3), initiator aa-tRNA, and the 30S subunit onto the start codon through interactions with the 30S Shine-Dalgarno (SD) RNA sequence, and ii) release of the initiation factors through the 50S subunit binding to the 30SIC. i) Initiation factors regulate translation initiation, requiring the initiator aa-tRNA, N-formylmethionine (fMet)-tRNAfMet, to be accommodated into the ribosomal P-site and decode the corresponding AUG start codon (Rodnina, 2018). The GTPase IF2 plays a large role in this selection, through direct contacts with fMet-tRNAfMet localized to the 3′-ACCAAC and the amino acid (Wu and RajBhandary, 1997; Guenneugues et al., 2000; Milon et al., 2010; Roy et al., 2017). ii) After accommodation, the 50S ribosomal subunit binds the 30SIC, catalyzing a series of conformational changes and ultimate release of initiation factors, to form the mature 70S initiator complex (70SIC) (Rodnina, 2018). As with many natural processes, there are exceptions to the general scheme of translation initiation including prokaryotic mRNA that lack a SD sequence, alternate starts codons (Tharp et al., 2020b) or the lack of a 5′UTR (Rodnina, 2018). However, the standard SD-led initiation at an AUG start codon, as described above, is almost exclusively explored with respect to initiator tRNA engineering.
Eukaryotic initiation resembles the path in prokaryotes with eIF1A, eIF2, and eIF1 homologous to bacterial IF1, IF2, and IF3, respectively (Brito Querido et al., 2024). In this process, the 43S pre-initiation complex (PIC) is formed through binding of initiation factors eIF1, eIF3, eIF1A, and eIF5, and a ternary complex [GTP-bound eIF2 complexed to the initiator aa-tRNA (Met-tRNAiMet)] to the 40S subunit (Lapointe et al., 2022). In eukaryotes, there is no SD sequence, therefore the 43S PIC is recruited to the 5′ end of an mRNA by the mRNA-bound eIF4F complex with the help of eIF4A and eIF4B to form the 48S PIC. The 48S PIC then scans the 5′UTR for a start codon (Wang J. et al., 2022). Upon recognition of this codon, most initiation factors are released, and the 60S subunit binds (catalyzed by eIF5B) to produce the 80SIC with a decoded AUG start codon in the ribosomal P-site (Brito Querido et al., 2024).
After translation initiation, the ribosome complex is ready to grow the nascent polypeptide chain. This is done through a well-conserved cyclic process to incorporate elongator tRNAs and form peptide bonds between amino acids (Dever et al., 2018). In prokaryotic systems, GTP-bound EF-Tu supports aa-tRNAs to the 70S ribosomal A/T-site where it decodes the exposed mRNA codon to promote tRNA accommodation into the ribosomal A-site and EF-Tu release. A peptide bond is then formed between the accommodated A-site and P-site aa-tRNAs (including de-acylation of the P-site tRNA) within the ribosomal peptidyl transferase center (PTC). Elongation factor P (EF-P) binds near the E-site to accommodate this reaction. Afterwards, the ribosome translocates along the mRNA by one codon (moving the tRNA species to the E- and P-sites, respectively), thereby opening the A-site to accept another aa-tRNA and repeat the elongation cycle (Xu et al., 2022). Elongation factor G (EF-G) binds near the A-site to disrupt codon-anticodon interactions and catalyze translocation (Liu et al., 2014; Rodnina et al., 2020). Finally, elongation factor thermostable (EF-Ts) catalyzes GDP-to-GTP nucleotide exchange to recycle the EF-Tu used (Gromadski et al., 2002; Kavaliauskas et al., 2012).
Eukaryotic elongation is similar to that of prokaryotes. GTP-bound eEF1A binds an aa-tRNA and is transported to the eukaryotic 80S ribosome (Dever et al., 2018). Base pairing interactions between the aa-tRNA anticodon and A-site codon catalyzes GTP hydrolysis of eEF1A (Shao et al., 2016), leading to its dissociation from the ribosome and aa-tRNA accommodation (Jobe et al., 2019). Peptide bond formation and translocation is similar to that of prokaryotic systems (Ben-Shem et al., 2011). eIF5a (bacterial EF-P ortholog) also binds the E-site and interacts with the acceptor arm of the peptidyl-tRNA to promote favorable substrate positioning and efficient peptide bond formation (Gutierrez et al., 2013; Melnikov et al., 2016b; Shin et al., 2017). Translocation is enhanced by eEF2, and eEF1B (orthologs of EF-G and EF-Ts, respectively) to catalyze a similar nucleotide exchange as EF-Ts to renew eEF1A for another cycle of elongation (Dever et al., 2018).
2 Acceptor domain engineering for improved EF-Tu interactions2.1 Basis of tRNA engineering for improved interactions with EF-TuNatural aa-tRNAs bind EF-Tu within a remarkably narrow affinity range which is necessary for unbiased transport of all amino acids to the ribosome and smooth mRNA translation (Louie et al., 1984; Ledoux and Uhlenbeck, 2008). This affinity is fine-tuned so that aa-tRNAs bind sufficiently to catalyze translation, but also dissociate at a rate fast enough to permit downstream peptide bond formation (Schrader et al., 2011; Ieong et al., 2014). Extensive research has determined the interface between EF-Tu and aa-tRNA (Figure 3A); importantly, the acceptor domain and amino acid moiety of aa-tRNAs are both responsible for binding EF-Tu (Kjeldgaard et al., 1993; Nissen et al., 1995; Nissen et al., 1996; Nissen et al., 1999; Sanderson and Uhlenbeck, 2007a; Schmeing et al., 2009; Kavaliauskas et al., 2012; Yikilmaz et al., 2014; Fischer et al., 2015; Johansen et al., 2018). Furthermore, both the tRNA sequence and amino acid structure work inversely together to achieve an affinity within this narrow range for optimal translation (Louie and Jurnak, 1985; LaRiviere et al., 2001; Asahara and Uhlenbeck, 2002; Asahara and Uhlenbeck, 2005; Dale et al., 2004; Dale and Uhlenbeck, 2005). Therefore, amino acids with weaker binding affinities are generally attached to tRNAs that have an acceptor domain with high affinity for EF-Tu and vice versa (Figure 3B). A series of mutagenesis-based experiments and computational modeling have further determined that base pairs 49:65, 50:64, and 51:63 within the T-stem determine the tRNA body’s affinity to EF-Tu (Figure 3C), irrespective of the other acceptor domain nucleotides within the interface of EF-Tu (Sanderson and Uhlenbeck, 2007b; Eargle et al., 2008; Schrader et al., 2009; Schrader and Uhlenbeck, 2011). According to this research, these three T-stem base pairs contribute to EF-Tu affinity independently of each other, suggesting that one can accurately predict a bacterial tRNA’s affinity for EF-Tu by summing the known contributions of the individual base pairs. Thus, engineering tRNA T-stems for increased EF-Tu affinity is a common, modular strategy to compensate for the incorporation of ncAAs that can be difficult ribosomal substrates (Figure 4).
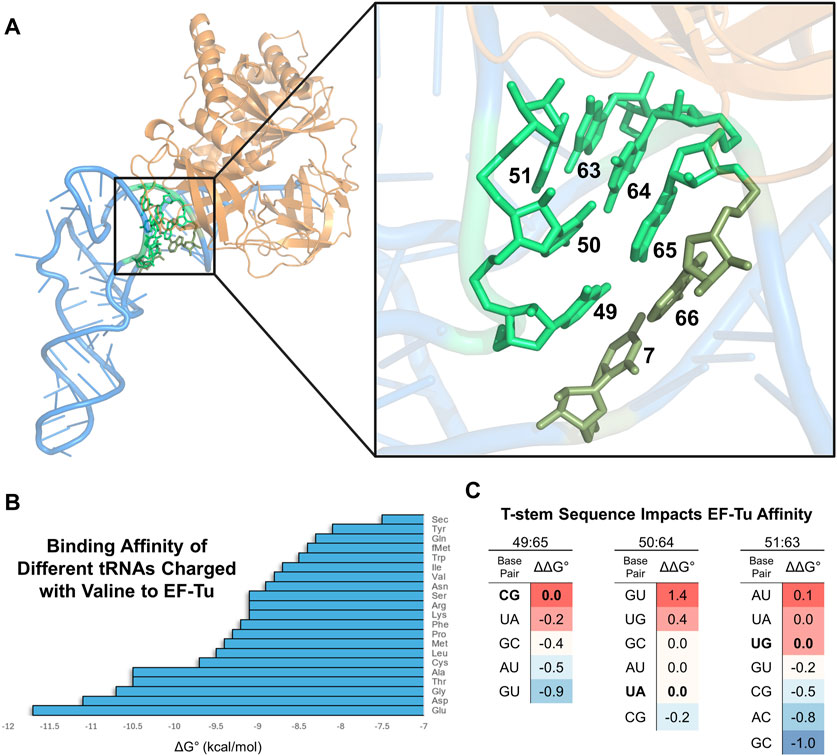
Figure 3. tRNA interacts with EF-Tu to moderate ribosome acceptance of aa-tRNAs. (A) Visualization of E. coli Cys-tRNACys (blue) bound to EF-Tu (orange) (PDB: 1B23). The black box highlights EF-Tu interactions with the acceptor domain of the tRNA. A zoom-in on the box shows the specific bases involved (highlighted in bright green). Base pair 7:66 (olive green) contributes least to the affinity, while the other three pairs contribute more. (B) Graph showing the different binding affinities of natural elongator valine-charged E. coli tRNAs to EF-Tu (Asahar and Uhlenbeck, 2002: Copyright (2002) National Academy of Science, United.States). (C) Different T-stem base pairs [highlighted in panels (A, B)] modularly impact aa-tRNA binding to EF-Tu. The bolded base pairs for each of the three T-stem base pairs represent the natural sequence when calculating the relative ΔΔG° to compare binding affinity differences. The intensity of red and blue shading indicates a base pair with relatively higher or lower affinity to EF-Tu at that position, respectively. Data from Schrader and Uhlenbeck, (2011) with E. coli tRNAPhe and figure repurposed from Shrader and Uhlenbeck, (2018).
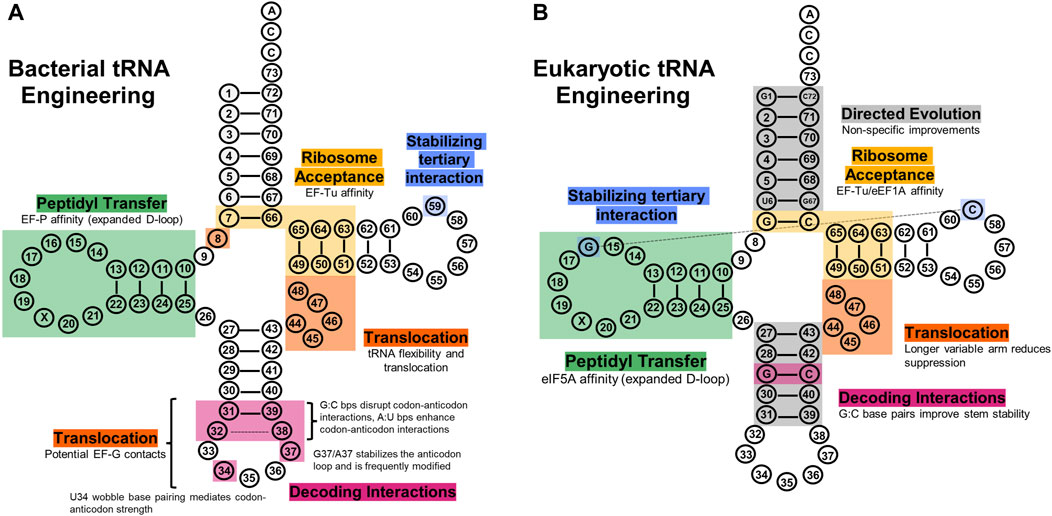
Figure 4. tRNA engineering sites explored to improve translational fidelity. (A) Bacterial tRNA regions have shown utility in improving various aspects of translation, such as ribosome acceptance (gold), decoding (pink), peptidyl transfer (green), translocation (orange), and tertiary structure (blue). (B) Eukaryotic tRNA regions engineered to improve their translational fidelity. Directed evolution experiments have elucidated the acceptor stem and anticodon stem as sites that non-specifically improve eukaryotic tRNA translational fidelity (grey), and other regions have been rationally engineered similarly to bacterial systems.
2.2 Examples of improving EF-Tu binding2.2.1 Canonical elongator tRNAsAs mentioned, canonical tRNAs have evolved to exhibit a wide range of affinities toward EF-Tu. Therefore, when engineering a tRNA for improved translational fidelity with a ncAA, it can be sufficient to use a natural tRNA with a high affinity for EF-Tu (e.g., tRNAGlu). This strategy has notably allowed for the implementation of d-amino acids (AAs), which are pharmacologically relevant, (Feng and Xu, 2016; Melchionna et al., 2016; Wang L. et al., 2022), but notoriously difficult ribosomal substrates (Blackmond, 2010; Fujino et al., 2013; Englander et al., 2015; Fleisher et al., 2018; Liljeruhm et al., 2019; Melnikov et al., 2019). In one case, charging tRNAGly (fourth strongest affinity to EF-Tu) with various d-AAs led to increased—but variable—single, double, and even triple consecutive insertions (Achenbach et al., 2015). A similar approach utilized a tRNAGlu (strongest natural EF-Tu affinity) variant (Terasaka et al., 2014) to efficiently incorporate up to 10 consecutive d-Ala or d-Ser amino acids into a peptide, and four or five consecutive d-AAs into a macrocyclic peptide in vitro, particularly useful for drug discovery (Katoh et al., 2017b; Goto and Suga, 2021). Although tRNA engineering alone demonstrated improved installation efficiency, optimization of IF2, EF-Tu, and EF-G concentrations further improved results—demonstrating the impact and complexity of other factors in cell-free protein synthesis.
2.2.2 Methanocaldoccocus jannaschii tRNATyr engineeringThe M. jannaschii (Mj) tyrosyl-tRNA synthetase (TyrRS):tRNATyr pair is commonly used for GCE in Escherichia coli (Xie and Schultz, 2006). After modifying loop regions of Mj tRNATyr to be orthogonal in E. coli (Wang and Schultz, 2001), further research sought to improve its elongation efficiency. Specific nucleotides in the acceptor domain (2:71, 3:70, 6:67, and 7:66 in the acceptor stem, and the entire T-stem) were selected based on a co-crystal structure of EF-Tu with Cys-tRNACys (Nissen et al., 1999) and subsequently randomized (Guo et al., 2009). Several effective suppressor tRNA variants emerged which improved the incorporation of ncAAs. In particular, tRNAoptCUA was only modified at base pairs 49:65, 50:64, and 51:63, and found to be capable of installing six ncAAs (Young et al., 2010). Notably, the incorporation efficiency differed depending on the ncAA, which holds true with previous comments about the amino acid contacts affecting affinity with EF-Tu.
A separate investigation used a directed evolution approach to improve the ribosomal efficiency for installation of three 3-halo-tyrosines (Maranhao and Ellington, 2017). These efforts focused on modifying the acceptor and T-stem nucleotides that interact with EF-Tu: nucleotides 3, 6, 68, and 71 in the acceptor stem, and base pairs 49:65, 50:64, and 51:63 in the T-stem. Five evolved tRNAs demonstrated 12 to 20-fold higher suppression efficiency for single incorporation of all 3-halo-Tyr variants. However, in the process of evolution, these tRNAs lost varying degrees of orthogonality in the cell, demonstrating that engineering one aspect of a tRNA can impact other tRNA functions.
2.2.3 tRNAPyl engineeringPyrrolysine (Pyl) is the 22nd proteinogenic amino acid found in archaea (Tharp et al., 2017) and some bacteria (Matassi, 2017) with its own incorporation machinery: pyrrolysyl-tRNA synthetase (PylRS) and its cognate tRNA (tRNAPyl). This system has been extensively researched for its use in GCE because of the promiscuity of the PylRS binding pocket and the orthogonality of the archaeal PylRS:tRNAPyl protein synthesis system in E. coli (Tharp et al., 2017) and eukaryotic systems (Chin, 2014; de la Torre and Chin, 2021). Significant research has focused on expanding the substrate capacity of PylRS and engineering PylRS:tRNAPyl pairs to be mutually orthogonal (Wan et al., 2014; Beattie et al., 2023; Gong et al., 2023). However, others have focused on optimizing tRNAPyl for improved translational fidelity (Wang J. et al., 2016). In one case, rational engineering of the T-stem and acceptor stem in Methanosarcinaea barkeri tRNAPyl resulted in improved binding to PylRS, and E. coli EF-Tu for incorporation of N(ε)-acetyl-l-lysine (AcK) (Fan et al., 2015). Small libraries of tRNA mutants were created at base pairs 2:71, 3:70, 6:67, and 7:66 in the acceptor stem, and 49:65 and 50:64 in the T-stem. It was found that changes to base pairs 7:66, 49:65, and 50:64, which are well-known EF-Tu affinity determinants (Asahara and Uhlenbeck, 2002), benefited from mutations. The best tRNA variant, tRNAPyl-opt, with G7:C66, U49:A65, and G50:C64 had a ∼3-fold improvement in AcK incorporation into sfGFP and human histone H3. tRNAPyl-opt was also used to incorporate two different ncAAs simultaneously at different sites within a protein (Zheng et al., 2018). These results collectively demonstrate the effectiveness of T-stem engineering to incorporate ncAAs. Combined with its orthogonality in heterologous systems, tRNAPyl is a responsive platform that can be fine-tuned to efficiently accommodate various ncAAs.
2.2.4 tRNASec engineeringSelenocysteine (Sec) is the 21st genetically encoded amino acid, found in all three domains of life, and can imbue proteins with unique properties such as overoxidation resistance, diselenide bond formation, and ability to be chemically modified (Arnér, 2010; Reich and Hondal, 2016; Mousa et al., 2017). Unlike the other natural amino acids, the mechanism for incorporating Sec into the ribosome involves additional steps and translation factors, including a specialized elongation factor, which makes site-specific Sec incorporation into proteins a formidable challenge (Yoshizawa and Böck, 2009; Chung and Krahn, 2022; Wright and O’Donoghue, 2024). The extended, 8 bp acceptor stem of tRNASec is a distinguishing feature that allows recognition by SelB and was originally hypothesized to prevent it from binding to EF-Tu (Baron and Böck, 1991). It was later shown that EF-Tu is capable of binding extended acceptor stems, albeit at a lower affinity, and rather it is the antideterminant sequence in tRNASec that specifically blocks EF-Tu binding (Rudinger et al., 1996). The antideterminant sequence in tRNASec consists of base pair C7:G66 in the acceptor stem and base pairs G49:U65 and C50:G64 in the T-stem—the same base pairs that are modified to improve EF-Tu affinity in other tRNAs. Thus, these base pairs act as a double-edged sword for EF-Tu binding; they can drastically improve and inhibit tRNA affinity for EF-Tu (Rudinger et al., 1996; Giegé and Eriani, 2023).
With this information, it suggests that tRNASec could be engineered to be recognized by EF-Tu instead of SelB. This would open the possibility for overexpression of selenoproteins and site-specific installation of Sec to harness its enhanced chemical properties. To achieve this, hybrid tRNAs were created between tRNASec and tRNASer (tRNAUTu, tRNASecUx, tRNAUTuX, and tRNAUTuT variants) by several groups. These hybrid tRNAs successfully demonstrated the possibility of EF-Tu-mediated Sec insertion (Aldag et al., 2013; Miller et al., 2015; Thyer et al., 2015; Fan et al., 2018).
The earliest example (tRNAUTu, Aldag et al., 2013) utilized tRNASer as the major scaffold; it retained the recognition elements for EF-Tu (G7:C66, C49:G65, and A50:U64), and incorporated the rest of the extended acceptor stem of tRNASec necessary for SelA recognition (to convert Ser-tRNASec to Sec-tRNASec). This created a 13 bp acceptor domain conducive for SelA recognition, but with EF-Tu identity elements already present in tRNASer to mimic the translation pathway of canonical AAs. Although a great leap forward in simplifying Sec’s translation pathway, this tRNA resulted in low selenoprotein yield and significant serine misincorporation (as EF-Tu still recognizes Ser-tRNAUTu) (Aldag et al., 2013). Optimizing Ser to Sec conversion became the focus of subsequent rational tRNA engineering efforts that were outside of the acceptor domain (tRNAUTuX, Miller et al., 2015).
Another group utilized E. coli tRNASec as a scaffold to design a tRNA with heightened EF-Tu binding affinity (Thyer et al., 2015). They accomplished this through tuning the antideterminant sequence (base pairs 7:66, 49:65, and 50:64) known to govern EF-Tu affinity (Rudinger et al., 1996; Schrader et al., 2009; Schrader and Uhlenbeck, 2011). Additionally, the authors hypothesized that tRNAUTu demonstrated Ser misincorporation due to reduced SelA contacts with the D- and T-loops of tRNASec as shown in the crystallized SelA structure (Itoh et al., 2013a; Thyer et al., 2015). Results indicated that G7:C66, U49:G65, and C50:U64 modifications on the tRNASec scaffold (tRNASecUx) greatly improved EF-Tu affinity and subsequent Sec insertion efficiency compared to tRNAUTu.
Later, another group re-engineered tRNAUTu by creating a small library of variants combining different tRNASer and tRNASec sequence segments. The segments tested were the acceptor stem, T-stem, and nucleotide 59 in the T-loop from either tRNASer or tRNASec. Nucleotid as tested because it is a conserved base (Romby et al., 1987; Lowe and Chan, 2016) relevant for maintaining 3D L-shape (Sanbonmatsu et al., 2005; Pan et al., 2008; Itoh et al., 2013b) and SelA binding (Fischer et al., 2016). This strategy resulted in many tRNA variants with modified T-stems, increasing their affinity to EF-Tu (Lajoie et al., 2013). However, the best performing tRNA (tRNAUTuT6) was formed by only changing A59C from the original tRNAUTu (Fan et al., 2018). This suggests that other T-arm nucleotides can contribute to translational fidelity, perhaps by improving interactions with SerRS, SelA, EF-Tu, and/or improved tertiary tRNA structure. The unique batch of tRNA variants from this study provided an unclear pattern on how to increase EF-Tu affinity.
Combined, these efforts demonstrate that Sec translation can be rewired for elongation by EF-Tu through focused engineering on the tRNA acceptor domain. However, the efficiency of these changes is limiting, most likely due to the fact that tRNASer and tRNASec have evolved for specific interactions with their translation machinery. Therefore, as discussed below, there are alternative strategies that can be taken to increase translation efficiency.
2.3 The curious case of EF-TsEF-Ts canonically function by binding EF-Tu-GDP and catalyzing GDP dissociation, allowing EF-Tu to rebind GTP and deliver another aa-tRNA to the ribosome (Miller and Weissbach, 1970; Weissbach et al., 1970; Miyajima and Kaziro, 1978; Pedersen et al., 1978; Romero et al., 1985; Wang et al., 1997). However, there is also research indicating that EF-Ts may have a more direct role in elongation by interacting directly with the aa-tRNA (Romero et al., 1985; Schwartzbach and Spremulli, 1991; Bubunenko et al., 1992) by i) regulating ternary complex formation and dissociation in response to cellular growth or stress signals, and ii) forming quaternary complexes that dramatically increase the rate of protein synthesis (Burnett et al., 2013; Burnett et al., 2014). Although there is no solved structure for aa-tRNA-EF-Tu-GTP-EF-Ts, it has been suggested that direct interactions between EF-Ts and the ternary complex are possible (Kawashima et al., 1996; Gromadski et al., 2002; Burnett et al., 2013) and may bind the aa-tRNA acceptor stem and elbow region in a conformation similar to the previously characterized E. coli EF-Tu-EF-Ts complex (Kawashima et al., 1996). It is unclear exactly how EF-Ts may interact with the complexed aa-tRNA, and whether tRNA engineering to improve favorable quaternary interactions with EF-Ts is a viable strategy for improving translational fidelity.
3 Improving codon-anticodon interactionsAfter the ternary complex has delivered aa-tRNA to the ribosome, the complex undergoes a series of interactions to ensure proper acceptance into the A-site for peptidyl transfer (Rodnina et al., 2017). Decoding efficiency essentially relies on a careful balance of codon-anticodon interaction strength and the ability of tRNA to adopt a proper conformation, both of which have been evolutionarily tuned, in part by tRNA modifications, to accommodate different codons and aa-tRNA combinations (Lorenz et al., 2017; Uhlenbeck and Schrader, 2018). The principles guiding this idiosyncratic tuning are less complete than the principles that have been shown to guide EF-Tu-related tRNA engineering efforts (Figure 4). Regardless, there has still been an exciting amount of information hinting at tRNA engineering principles to help improve their decoding mechanics, focused overwhelmingly on bacterial systems.
3.1 Engineering the anticodon arm for improved translational fidelityWhile the anticodon sequence itself determines a substantial proportion of the codon-anticodon interactions within the ribosome (Rozov et al., 2016; Loveland et al., 2017; Rodnina, 2023), it is also well-known that surrounding nucleotides and base pairs within the anticodon arm influence these dynamics to uniformly tune the different strengths of codon-anticodon interactions to ensure uniform decoding efficiency and rate of mRNA translation (Olejniczak et al., 2005; Olejniczak and Uhlenbeck, 2006; Ledoux and Uhlenbeck, 2008). This covariance between the anticodon sequence and other anticodon arm elements is known as the “extended anticodon” (Yarus, 1982; Yarus et al., 1986; Raftery and Yarus, 1987; Kleina et al., 1990; Auffinger and Westhof, 2001; Murakami et al., 2009; Schmeing et al., 2011; Shepotinovskaya and Uhlenbeck, 2013; Cervettini et al., 2020). Specifically, it has been shown that the identities of positions 32, 37, 38, and at least two base pairs in the anticodon stem are well-conserved (Saks and Conery, 2007).
The flexibility of the anticodon stem-loop, largely influenced by base pair 31:39 and non-canonical interactions between nucleotide 32:38 (Auffinger and Westhof, 1999), impacts this decoding efficiency and codon recognition specificity (Olejniczak et al., 2005; Ledoux and Uhlenbeck, 2008; Sigal et al., 2024). “Stiffer” anticodon arms result from G:C base pairs at these positions, generally distorting the canonical anticodon loop conformation and disrupting the binding of GC-rich codons (Olejniczak et al., 2005; Olejniczak and Uhlenbeck, 2006; Murakami et al., 2009), whereas weaker interactions, including the rare A32:U38 interaction, allows easier codon-anticodon base pairing (Ledoux et al., 2009; Schmeing et al., 2011; Grosjean and Westhof, 2016). Therefore, tRNA sequences are seemingly tuned for optimal bending in the ribosome to off-set the strengths of different codon-anticodon interactions (Shepotinovskaya and Uhlenbeck, 2013).
A more recent example (Katoh and Suga, 2024) leveraged these principles to further improve the translational efficiency of an engineered tRNA platform (discussed below) to incorporate especially difficult ribosomal substrates that had eluded previous tRNA engineering efforts (Lee et al., 2022). Crucially, screening the anticodon stem of the scaffold tRNAPro1E2 (Katoh et al., 2017a) led to different tRNAPro1E2 variants each uniquely optimized to one of five different anticodon sequences. Then, anticodon loop mutations at positions 32, 33, 37, and 38 were introduced according to the native E. coli anticodon sequences leading to further improvement of ncAA incorporation efficiency. Combined, these improvements allowed for multi-site incorporation of βAAs at five different codons in vitro and the first reported ribosomal installation of 10 consecutive β-homophenylglycine (βPhg) residues. The G:C or C:G base pair at 31:39 was conserved across all codons, along with a conserved A37.
However, it is worth noting that mutational, structural, and bioinformatic has also identified positions dispersed throughout all arms of bacterial tRNAs that influence proper bending and decoding specificity (Saks and Conery, 2007; Schmeing et al., 2011; Shepotinovskaya and Uhlenbeck, 2013). In this way, the “extended anticodon” truly extends throughout the entire tRNA, potentially complicating tRNA engineering efforts when focusing on multiple tRNA regions.
3.2 Anticodon arm modifications influence decodingtRNAs are frequently post-transcriptionally imbued with diverse modifications that drastically impact their structure and function (Lorenz et al., 2017; Suzuki, 2021; Cappannini et al., 2024; Yared et al., 2024). tRNAs exhibit diverse modifications in all arms, but they are most frequent at the tRNA core and anticodon arm (Cantara et al., 2011; Smith et al., 2024). Anticodon arm modifications in particular have thus been targets for uncovering tRNA translation fidelity principles as an “unrealized genetic code” to complement the tRNA sequence itself (Agris et al., 2017). In particular, modifications at positions 32, 34, and 37 are frequent and chemically varied, especially at the first two positions (Jühling et al., 2009).
The most well-known examples of post-transcriptional modifications impacting decoding are those that occur at position 34, the “wobble” base (Crick, 1966; Agris, 1991; Demeshkina et al., 2012). In some cases, the modification status at position 34 expands wobble pairing to all four mRNA nucleotides at the third codon position (Rogalski et al., 2008; Bloom-Ackermann et al., 2014). Other modifications have been shown to impact decoding specificity or even completely disable codon binding altogether (Kurata et al., 2008; Kumbhar et al., 2013; Nedialkova and Leidel, 2015; Nakano et al., 2016; Song et al., 2022). A34 is also almost universally modified to inosine to expand decoding potential to C, U, and A (Crick, 1966). Position 37 does not directly interact with the codon, but is often a purine frequently modified to stabilize Watson-Crick base pairing at the first two positions of a codon (Väre et al., 2017). Moreover, position 32 modifications generally help to create non-canonical interactions at base pair 32:38 to stabilize the U-turn structural motif of the anticodon arm relevant for ribosomal A-site acceptance and codon discrimination (Auffinger and Westhof, 1999; Cantara et al., 2012; Väre et al., 2017). In addition to the anticodon loop, tRNAs are also often simultaneously modified at anticodon arm positions. These modifications are connected with the anticodon sequence, synergistically functioning to alter tRNA structure and decoding efficiency (Nishimuka, 1972; Jühling et al., 2009; Cantara et al., 2012; Grosjean and Westhof, 2016; Väre et al., 2017). However, the exact impact of adding or removing a modification across different tRNAs is not well understood, and some unmodified tRNAs can still function (Phizicky and Alfonzo, 2010; Lorenz et al., 2017). One heuristic approximation is that GC-rich decoding tRNAs are modified to reduce binding affinity whereas AU-rich decoding tRNAs are modified to improve codon binding (Uhlenbeck and Schrader, 2018; Sigal et al., 2024). One recent example improved the UAG decoding efficiency of a previously engineered opttRNACUATyr (Young et al., 2010) by replacing adenine with 2, 6-diaminopurine (D) at positions 31 and 36 (Mala and Saraogi, 2022). D is similar to adenine but has an added C2 amino group, enabling the formation of an additional hydrogen bond in the D-U base pair compared to A-U pairing, which strengthens codon-anticodon interactions and stabilizes RNA duplexes (Strobel et al., 1994).
It has also been hypothesized that the overreliance on synthetic tRNAs that lack post-transcriptional modifications limits tRNA activity (McFeely et al., 2022); it was recently shown that wild-type (i.e., fully modified) tRNAs significantly outcompeted their synthetic counterparts (3 to 4-fold improvement) for the same cognate sense codons (McFeely et al., 2022). Interestingly, heterologous tRNAs such as tRNAPyl-opt can also be engineered to reduce dependence on post-transcriptional modifications and improve suppression efficiency (Baldridge et al., 2018).
Combined, tRNA modification engineering will almost certainly aid in producing more effective synthetic tRNAs, although more research is needed to better define fundamental principles.
3.3 Engineered quadruplet codons efficiently expand the genetic codeSignificant research efforts have focused on engineering triplet codons for improved translational fidelity. However, there are limitations for genetic code expansion because triplet codons result in only 64 possible codon combinations, which traditionally encode for 20 proteinogenic AAs (plus Sec and Pyl) and three stop codons. Therefore, there has been significant research efforts toward engineering quadruplet codon-decoding tRNAs—tRNAs that decode four base codons on the mRNA—to vastly expand the available number of empty codons (up to 256 free codons) (Figure 5). Efficient quadruplet codons are also inherently orthogonal to the 64 triplet codons, thereby not requiring codon reassignment.
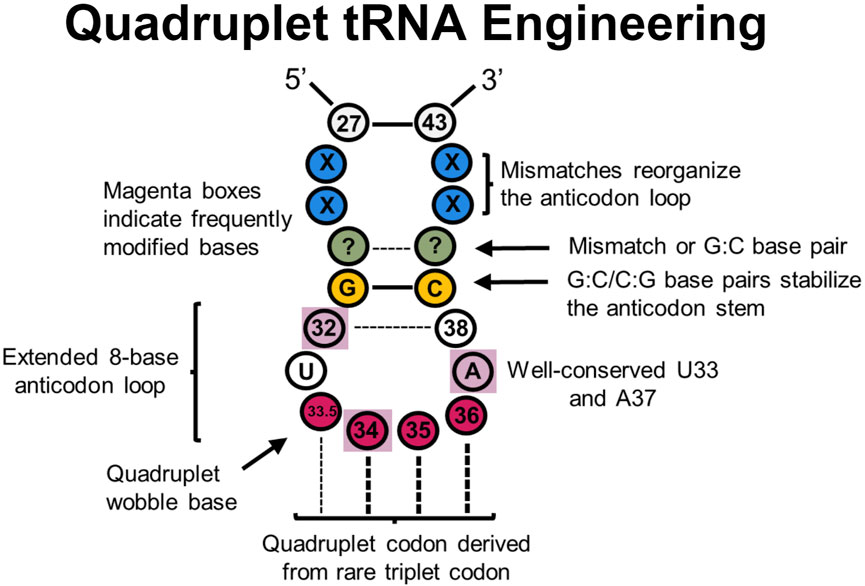
Figure 5. tRNA sites explored in quadruplet tRNAs to improve translational fidelity. Efficient quadruplet tRNAs have a well-conserved G:C/C:G base pair adjacent to the anticodon loop (yellow), A37 and U33 residues, and base pair mismatches near the beginning of the anticodon stem (blue) which adjusts the conformation of tRNAs. Frequently post-transcriptionally modified residues (purple boxes) also impact translational fidelity, although position 33.5 base pairing with a quadruplet codon is not necessary.
Such quadruplet decoding, or “programmed frameshifts,” occur in nature, often with an extended 8-base anticodon loop as opposed to the canonical 7-base loop (Riyasaty and Atkins, 1968; Riddle and Roth, 1970; Yourno and Kohno, 1972; Riddle and Carbon, 1973; Bossi and Smith, 1984; Farabaugh, 1996; Moore et al., 2000a; Walker and Fredrick, 2006; Atkins and Björk, 2009; Mohanta et al., 2023). Many experiments have since demonstrated that inserting a single nucleotide into the anticodon loop of a tRNA to create an 8-base anticodon results in moderate quadruplet decoding in vitro and in vivo, especially in combination with engineered synthetases (Curran and Yarus, 1987; Ma et al., 1993; Hohsaka et al., 1996; Hohsaka et al., 1999; Hohsaka et al., 2001; Moore et al., 2000b; Magliery et al., 2001; O’Connor, 2002; Ohtsuki et al., 2005; Taki et al., 2006; Wan et al., 2010; Chatterjee et al., 2012; Hankore et al., 2019; Dunkelmann et al., 2020; Dunkelmann et al., 2021; DeBenedictis et al., 2022). In a recent report, it was determined that nearly a third of the 20 bacterial tRNA isoacceptors can demonstrate modest quadruplet decoding activity after carrying the insertion of a single nucleotide insertion in the anticodon loop in E. coli (DeBenedictis et al., 2022). Additionally, while experiments with quadruplet M. barkeri tRNAPyl using a 7-base anticodon loop were unsuccessful, expanding them to 8-bases provided an avenue for quadruplet decoding (Wang N. et al., 2016). One potential issue with anticodon arm engineering is that the anticodon loop can be an essential identity element for binding respective aaRSs (Melnikov and Söll, 2019). Therefore, engineering the anticodon arm for improved decoding mechanics can also alter aminoacylation specificity and efficiency. Many engineered quadruplet tRNAs thus originate from tRNAPyl scaffolds as PylRS is known not to interact with the anticodon loop (Nozawa et al., 2009).
Designing quadruplet anticodon sequences derived from rarely-used triplet codons can help with endogenous machinery competing to perform triplet decoding as a general quadruplet tRNA engineering principle (Guo and Niu, 2022). In one case, a GGGU quadruplet tRNA was outcompeted by inserting Gly at the CCC codon in Xenopus oocytes (Shafer et al., 2004). In contrast, deriving quadruplet tRNAs from rarely used three base codon sequences in E. coli found more success in quadruplet decoding efficiency (Hohsaka et al., 2001). Interestingly, an in vivo library of E. coli tRNASer with randomized 8- or 9-base anticodon loops containing various quadruplet codons (NNNN) found that the most efficient tRNAs decoded CCCU, AGGA, UAGA, and CUAG codons—all of which are derived from rare triplet codons (Magliery et al., 2001). This strategy has also served to successfully incorporate ncAAs in E. coli (Anderson et al., 2004; Chatterjee et al., 2014).
As the anticodon arm (and entire tRNA, to an extent) often coevolves with its anticodon sequence, further modification of the anticodon arm outside of the quadruplet anticodon has been shown to improve decoding efficiency. In E. coli and mammalian cells alike, in vitro evolution of the entire tRNA anticodon arm generated mutants with efficient incorporation of ncAAs at the AGGA codon (Niu et al., 2013). Importantly, sequence convergence of base pairs G28:C36 and G27:C37, along with some base pair mismatch at 26:38 was observed. Combined, this sequence was thought to restructure the anticodon arm to help with A-site binding and translocation. Another investigation (Wang et al., 2014) evolved a series of M. barkeri tRNAPyl mutants for efficient quadruplet decoding with ncAAs using an engineered ribosome (ribo-Q1) (Neumann et al., 2010). tRNAs were randomized at positions 29–33 and 37–41 for four quadruplet codons (AGGA, AGTA, TAGA, and CTAG). The most efficient mutants all shared a G:C or C:G base pair adjacent to the anticodon loop, even though the size of the anticodon loop varied. Position A37 was also conserved among all efficient mutants, which is associated with reading frame maintenance (Agris, 2004). Follow up research varied the same regions in UAGN-decoding M. mazei tRNAPyl to improve quadruplet decoding efficiency and uncover a better mechanistic understanding of quadruplet decoding in E. coli (Wang N. et al., 2016). This led to significant quadruplet decoding efficiency improvements and incorporation of ncAAs with two PylRS variants, though quadruplet codon orthogonality was not obtained. Most strikingly, all observed tRNA mutants contained U33.5 regardless of the UAGN codon they decode, validating the previously hypothesized idea that strict Watson-Crick base pairing at all four positions is not required for quadruplet decoding (Guo and Niu, 2022). It is possible that U33.5 impairs interactions at position 32:38 to expand the anticodon loop and permit quadruplet decoding (Maehigashi et al., 2014; Wang N. et al., 2016) and/or preserves an essential U-turn structure in the anticodon loop necessary for translocation (Phelps et al., 2002; Phelps and Joseph, 2005). Importantly, all mutants contained A31G and U39C mutations, resulting in a new G:C base pair which strengthens the anticodon stem, but most mutants did not have Watson-Crick base pairing at pairs 29:41 or 30:40 which may restructure the anticodon arm for more efficient quadruplet decoding. It was also noted that position 33 and 37 were never mutated. While not specifically evolved for mammalian cells, these quadruplet tRNAs were shown to be effective in incorporating a ncAA in HEK 293T cells, suggesting that anticodon arm engineering strategies are applicable to a wide range of systems (Chen et al., 2018). Additional research (Mills et al., 2021) screened examples of these previously-engineered quadruplet-decoding tRNAPyl variants (Niu et al., 2013; Wang et al., 2014; Wang et al., 2016 N.; Chen et al., 2018) in HEK293 cells, and found that two of the tRNA variants (one evolved for mammalian cells, one with a simple quadruplet anticodon change) were suitable for use in ncAA implementation. Notably, engineered quadruplet tRNAs across all experiments were generally most effective only in the organism that were designed in.
A more recent investigation sought to improve different quadruplet E. coli tRNA suppression efficiencies through directed evolution (DeBenedictis et al., 2021) at the UAGA-codon, permitting multiplex decoding of four distinct quadruplet codons within a single reporter protein. It was found that mutations in the anticodon loop (positions 32 and 38), variable arm, central loop, D-stem, and acceptor stem improved quadruplet coding efficiency up to 80-fold. However, there was no clear consensus for these mutations. In a follow up experiment, focused libraries at positions 32, 37, and 38 further improved translation efficiency for different quadruplet codons in E. coli (DeBenedictis et al., 2022). Individual libraries for specific codons demonstrated enrichment for a specific nucleotide at one or more positions, suggesting significant covariance between the anticodon sequence and other regions of the anticodon arm. Additionally, there was a pronounced preference for base A37 across all codon libraries.
Quadruplet decoding has also been utilized to incorporate ncAAs in animals; to adapt quadruplet decoding ncAA incorporation in C. elegans, hybrid tRNAs containing the UCUA anticodon were created and demonstrated significant improvement in ncAA incorporation efficiency. They used M. mazei tRNAPyl variants already known to interact well with C. elegans translational machinery in triplet codon ncAA incorporation (Serfling et al., 2018; Davis et al., 2021) as a starting scaffold while overlaying anticodon arm modifications previous
留言 (0)