Bacterial strains and growth conditions
Clinical specimens suspected to contain Klebsiella isolates were collected from the sputum of patients at Zagazig University Hospital, Sharqia, Egypt, following the acquisition of informed consent. The collected samples were promptly transported to the laboratory for further processing. To ensure the safety of laboratory personnel and prevent cross-contamination, the samples were handled in accordance with standard biosafety protocols. Laboratory personnel wore appropriate personal protective equipment (PPE), including gloves and laboratory coats, during sample processing.
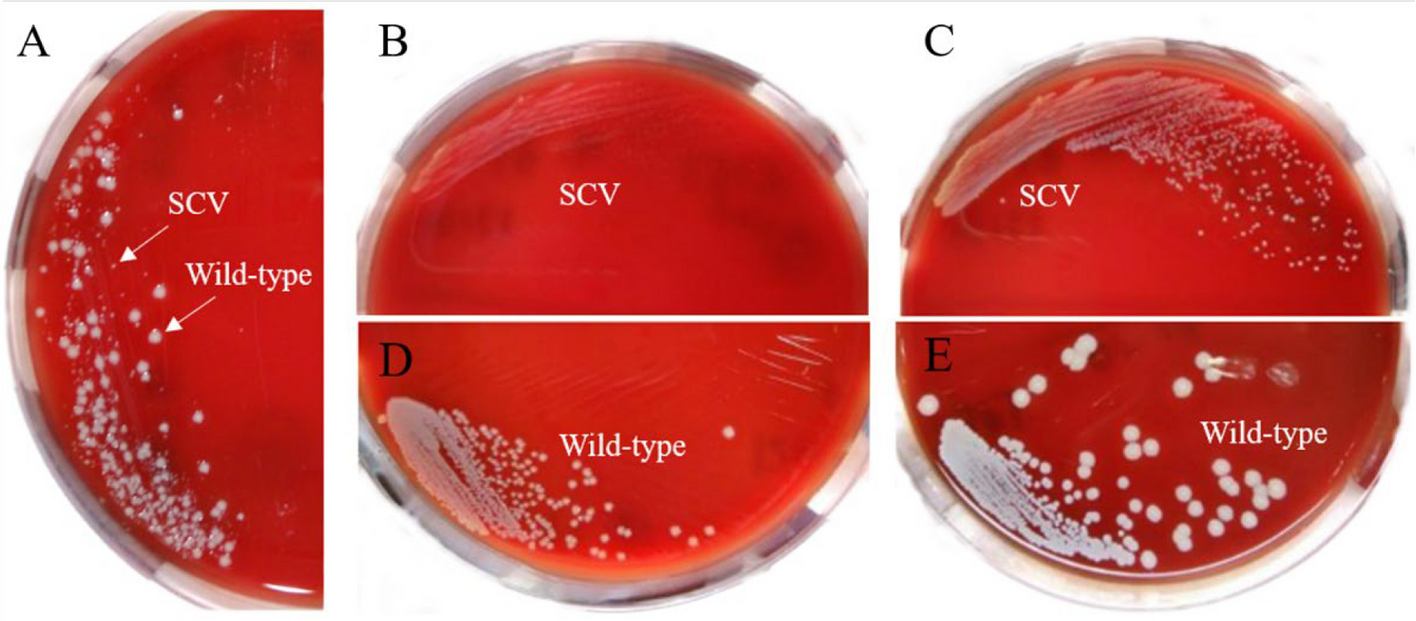
Serial dilutions of the collected samples were performed to achieve a suitable bacterial load for isolation. Using a sterile loop or spreader, the diluted samples were streaked onto selective agar plates specifically designed for the isolation of Klebsiella, such as MacConkey agar or blood agar (Oxoid, United Kingdom) [16]. These plates were then incubated at the optimal temperature for Klebsiella growth, typically 35–37°C, for a period of 24–48 h. Following incubation, the agar plates were examined for the presence of bacterial colonies. Colonies displaying characteristic Klebsiella morphology, such as a mucoid appearance, lactose fermentation, and non-hemolytic on blood agar plates, was selected. Using a sterile loop, these selected colonies were streaked onto fresh agar plates to obtain pure cultures, ensuring the isolation of individual Klebsiella strains [17]. The isolated colonies were then incubated under appropriate conditions to facilitate further growth and identification. To confirm the identity of the isolated Klebsiella strains, additional tests were conducted. Specifically, DNA extraction was conducted using the QIAamp DNA Mini Kit supplied from QIAGEN (USA) with Catalogue no. 51304. Subsequently, 16S rRNA sequencing was performed using the ready reaction Bigdye Terminator V3.1 cycle sequencing kit (Perkin-Elmer/Applied Biosystems, Foster City, CA), with Cat. No. 4336817, provided by Sigma Company, located in Giza, Egypt, for the accurate identification of the Klebsiella strains [18]. The GenBank accession number link provided: https://www.ncbi.nlm.nih.gov/nucleotide/OP942216.1.
Antibiotic susceptibility test
The antibiotic susceptibility test was conducted on the clinical isolates to determine the susceptibility pattern of bacteria. The antibiotic sensitivity test was conducted using the Kirby-Bauer method (also called the disc diffusion test) on Muller-Hinton agar [12]. Sterilized Müller-Hinton agar (Oxoid, USA) was poured into Petri dishes in 10 mL aliquots. Once the agar solidified, 100 µL aliquots of broth cultures, inoculated with 108 CFU mL−1 of the tested bacterial strains, were evenly spread over the surface of the agar plates. Antibiotic discs were carefully placed on the plate surfaces. Fifteen bacterial isolates were tested against the following antibiotics: Imipenem (IPM) (10 μg), Polymyxin B (PB) (300 μg), Amikacin (AK) (30 μg), Erythromycin (E) (15 μg), Trimethoprim/sulphamethoxazole (SXT) (25 μg), Cefoxitin (FOX) (30μg), Cefepime (FEP) (30μg), Ceftriaxone (CRO) (30μg), Ceftazidime (CAZ) (30μg), Gentamicin (CN) (10μg), Ciprofloxacin (CIP) (5μg), Erythromycin (E) (15μg), Chloramphenicol (C) (30mcg), Piperacillin (PRL) (100 μg), and Tetracycline (TE) (30 μg). The plates were then incubated upside down at 37°C for 16–18 h. The diameter of the inhibition zone around each antibiotic disc was measured in millimeters (mm). The interpretation of the inhibition zone diameter was classified as sensitive (S), intermediate (I), or resistant (R) based on the interpretative criteria recommended by CLSI [19] for antimicrobial susceptibility testing. Additionally, the multiple antibiotic resistances (MAR) index was calculated to determine the level of antibiotic resistance. The MAR index is the ratio between the number of antibiotics to which the bacteria are resistant and the total number of antibiotics used, as described by Sayah et al. [20].
Enrichment and Isolation of K. pneumoniae bacteriophage
Bacteriophages were isolated from different sewage water samples obtained from Sharkia Governorate, Egypt, using the enrichment technique [21]. Initially, 100 mL of sewage was filtered through a 0.45μm-filter membrane and combined with an equal volume of nutrient broth in 500 mL Erlenmeyer flasks. Subsequently, 5 mL of fresh K. pneumoniae culture (2.0 × 108 CFU mL−1) was added to each sewage sample. The flasks were incubated on a shaker (120 rpm) at 37 ºC for 24 h. After incubation, the mixture was centrifuged at 10,000 rpm for 20 min, and the resulting supernatant was filtered through a 0.45μm-filter membrane to detect the presence of phages using spot test and plaque assay methods with K. pneumoniae as the host. Phage titration was performed by tenfold serial dilution of the samples in saline solution and spotting them on lawns of K. pneumoniae, with the phage titer expressed in plaque forming units (PFU) per mL.
Purification of K. pneumoniae bacteriophage
Phages were propagated and purified from various single-plaque isolates following Kim et al. [21]. The isolated phages underwent five successive single-plaque isolations until homogenous plaques were obtained. In each isolation, a single plaque was picked and incubated with 1 mL of nutrient broth containing an overnight culture of K. pneumoniae at 37 ºC with agitation. After incubation, the phage-host mixture was centrifuged at 10,000 rpm for 10 min, and the supernatants were filtered through a 0.45μm Millipore filter to eliminate any bacterial contamination. The purified phages were stored at 4ºC for further characterization.
Electron microscopy analysis of isolated bacteriophage
The morphology and structure of the isolated bacteriophage were analyzed using transmission electron microscopy (TEM), according to the method described by Abdel-Haliem and Askora [22].The phage suspension was prepared, containing a concentration of 108 (PFU mL−1). A small volume of the phage suspension was applied onto Carbon-coated formvar films on 200 mesh copper grids. Excess liquid was carefully removed from the grid using filter paper. A few drops of the Sodium phosphotungstate solution were added onto the grid, covering the phage particles. Excess Sodium phosphotungstate solution was gently removed from the grid using filter paper. Images of the phage particles were captured using a Hitachi H600A electron microscope at the Faculty of Agriculture, Mansoura University, Egypt.
The one-step growth
The one-step growth curve of the phage was determined following a standard protocol [16]. Briefly, a culture of the host bacterium K. pneumoniae was grown to the logarithmic phase of growth. The bacterial culture was then infected with the phage at a multiplicity of infection (MOI) of 0.01, which means that for every bacterium, only one phage was added. After allowing the phage to adsorb to the bacterial cells for a specific duration 5 min, the mixture was diluted and plated onto agar plates to determine the number of viable phages (PFU) at time zero. This represented the initial phage count. Subsequently, samples were taken at regular intervals every 5 min for certain duration 2 h. Each sample was immediately diluted and plated onto agar plates to quantify the number of infective phages at each time point. This allowed for the construction of the growth curve. To calculate the burst size, the number of new phages released from each infected bacterium was determined by comparing the phage count at the peak of the growth curve with the initial phage count. The burst size represents the average number of phages released per infected bacterium. The experiment was performed in triplicate to ensure the reliability of the results, and the average values along with standard deviations were reported.
Host range
The host range of Kzag1 phage was determined experimentally using a collection of bacterial strains, including K. pneumoniae and related species, as potential hosts. Each bacterial strain was cultured to the logarithmic growth phase, and 100 µL of each culture was spread on separate agar plates. After drying, small drops of the phage suspension were added to the agar surface to allow phage attachment to the bacterial cells. The plates were then incubated at the optimal growth temperature for the bacterial strains at 37°C. After an appropriate incubation period 18–24 h, the plates were inspected for the presence of plaques, indicating successful phage infection and subsequent bacterial cell lysis. The absence of plaques indicated that the phage could not infect the specific bacterial strain.
The influence of temperature and pH on the stability of the phage
The influence of temperature and pH on the stability of the phage was examined. Thermal stability tests were performed following the protocol described by Mahmoud et al. [23]. Phage samples with a known titer (106–108 PFU mL−1) were exposed to different temperatures ranging from 30°C to 100°C for 10-min intervals in a water bath incubator. The infectivity of the phages was determined immediately after incubation using the double-layer agar plate method. Additionally, pH stability tests were conducted by inoculating a known phage suspension (106–108 PFU mL−1) into LB liquid medium obtained from Thermo Fisher Scientific, United States with pH values ranging from 3.0 to 12.0, followed by overnight incubation at 4°C. The viability of the bacteriophages was assessed using the overlay method as described by Adams [21].
The impact of the Kzag1 bacteriophage on the proliferation of K. pneumoniae
The impact of Kzag1 phage on the growth of K. pneumoniae was investigated. A culture of K. pneumoniae was diluted to a final density of 2.0 × 108 CFU mL−1and placed in a nutrient broth, then incubated at 37°C for 24 h. The phage suspension was also diluted to achieve a MOI of 0.1. Subsequently, 100 µL of various phage concentrations was added to the bacterial suspension [24], and the mixture was incubated under sterile conditions. The survival of K. pneumoniae was assessed at intervals of 5, 10, 15, 20, 25, 30, and 35 min using the plaque assay method.
The impact of individual Zag1 phage on biofilm formation
To assess the impact of Zag1 phage on biofilm formation by Klebsiella, the following experimental procedures were conducted according to Jamal et al. [25]. A biofilm was developed by introducing 200 μL of bacterial culture (108 CFU mL−1) into each well of a 96-well flat-bottomed polystyrene microtiter plate, followed by incubation at 37°C for 24 h with gentle agitation at 120 rpm. After the biofilm formation period, any excess fluid was discarded, and the wells were washed twice with 0.9% NaCl to eliminate unattached planktonic cells. The plate was then allowed to dry at 37°C for one hour. The isolated phages were diluted in 0.9% NaCl and added to the respective wells containing their host bacteria. Different concentrations of the Zag1phage was used, including MOI = 0.1, MOI = 1, and MOI = 5. The control wells were set up using uninoculated normal saline, and they remained untreated throughout the experiment. Specifically, the uninoculated normal saline serves as the negative control, while the well inoculated with bacterial culture serves as the positive control. The plates were incubated at 37°C with constant shaking at 120 rpm for an additional day after the establishment of the biofilm. Excess fluid was removed from each well, and the biofilms were washed as previously described. The wells were left to dry for one hour at 37°C. To determine the total biomass of the biofilm, staining with 1% crystal violet was performed for 20 min. The plates were then washed with distilled water and air-dried. Next, 200 μl of 0.9% NaCl solution was added to each well, and the absorbance was measured at OD570 using an ELISA plate reader (Biotek Synergy HT Microplate Reader, USA. Triplicate measurements were taken for both control and test samples.
Phage DNA sequencing and subsequent bioinformatics analysis
The phage DNA sequencing and subsequent bioinformatics analysis were carried out as follows: Initially, phage DNA was extracted using the phenol–chloroform method, following the protocol by Sambrook and Russell [26]. The purified phage DNA was then subjected to sequencing using the Illumina Miseq platform (Illumina, San Diego, CA, United States). The obtained sequencing data was assembled using SPAdeS v.3.13.0 [27] by Sanigen Inc., South Korea. To identify ORFs, a combination of Glimmer3 [28], GeneMarkS [29], and the RAST annotation server [30] was utilized. The annotated data were organized using Artemis [31]. Furthermore, the tRNA sequence within the phage genome was analyzed using the tRNAscan-SE program. Predictions for the functions of the phage proteins were made using NCBI BLASTp and the InterProscan program [32]. The annotated genome sequence of the KZag1 phage was deposited in the NCBI GenBank database under accession number OR502445. Phylogenetic analysis of the Zag1 phage was conducted by querying the Blast database and reconstructing a phylogenetic tree. Genomic sequences of relevant phages were retrieved from the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) [32]. These sequences underwent alignment using Clustal Omega to ensure precise alignment, considering sequence homology and structural similarities [33]. Subsequently, the aligned sequences were used to construct phylogenetic tree employing the robust maximum likelihood (ML) methodology [34]. Statistical analyses were conducted to evaluate the significance of the inferred phylogenetic relationships.
Statistical analysis
Each experiment was conducted in triplicate, and the average of the triplicate determinations was taken to represent the results. Statistical analyses were carried out using SPSS software package version 11.5 and Microsoft Excel 2010. The data were subjected to analysis of variance, and significant differences (p > 0.05) between means were determined according to Pallant [35].









留言 (0)