
記住我





An accurate selection of virtual patients is essential to ensure the reproducibility, accuracy, and breadth of information that a virtual clinical trial can provide. In practice, each virtual patient represents a set of unique input parameters and initial conditions used to simulate the model. These are usually determined via sampling from calibrated (or assumed) distributions or are obtained from pre-treatment simulations. Ideally a substantial virtual patient cohort is capable of representing both individual patients and population response.
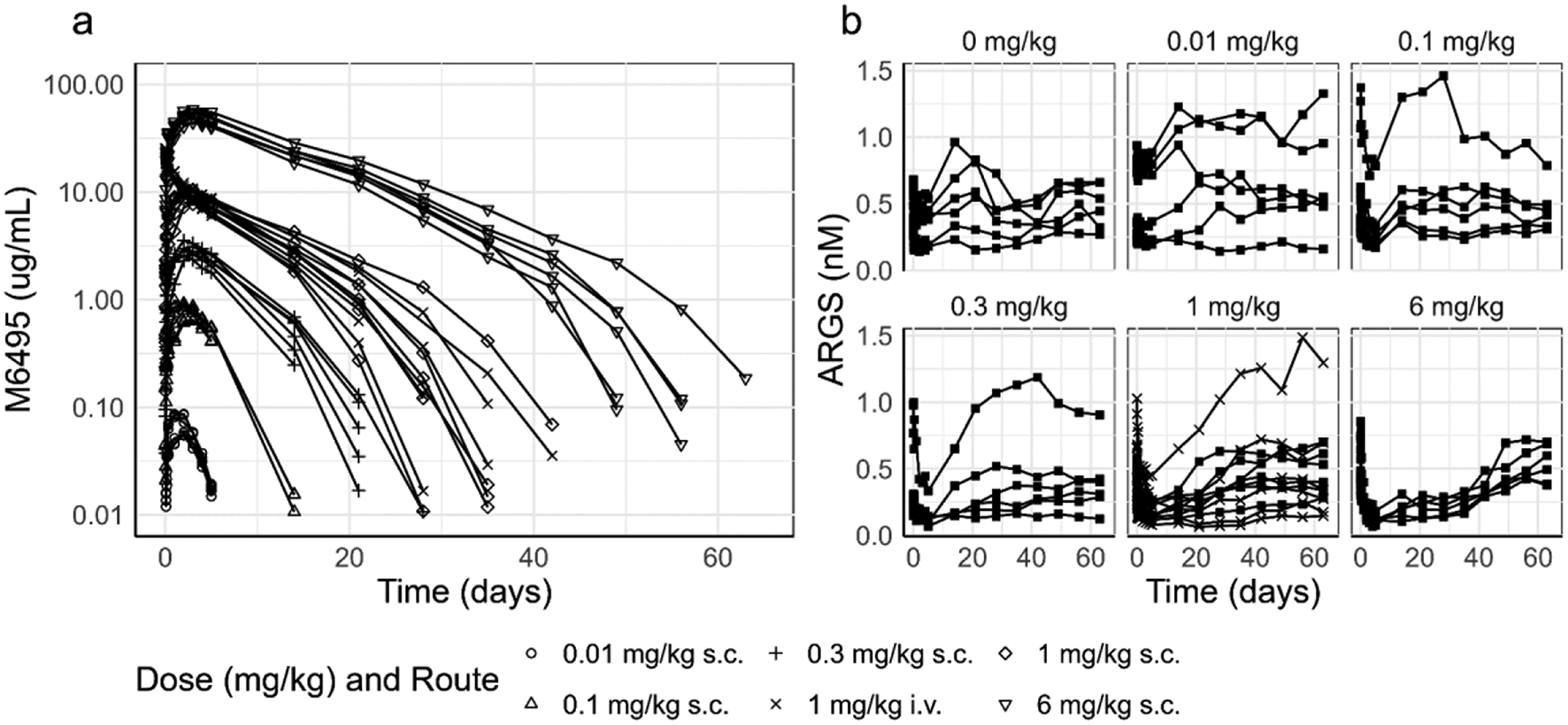
In this work, we implement the virtual patient selection method described in Fig. 1a (see Supplementary information for additional details). This method is adapted from [10, 12]. As the first step, the input parameters are randomly selected from estimated distributions via Latin Hypercube Sampling [13, 14]. We designate these patients as “proposed patients” and proceed by simulating their tumor growth from a few cells for 8000 days, using the QSP-IO model described in Fig. 1b; note that the 8000 day interval is a computational parameter and does not necessarily correspond to the real tumor growth duration. We also want to emphasize that this initial condition calculation is of negligible complexity compared to the virtual clinical trial simulation. We then select the patients that have plausible tumor size profiles, and we designate them as “plausible patients.” The initial condition (i.e., pre-treatment condition), in terms of all the cell and cytokine densities, of the plausible patients is set as the timepoint when the tumor reaches the desired initial tumor diameter (i.e., pre-treatment tumor size). Using these initial conditions, we construct three immune subset ratios shown in Fig. 2a and defined as:
$$ = \left\}&/\left( + } \right) > 1 - \varepsilon } \\ }} + }}}&/\left( + } \right) \leqslant 1 - \varepsilon } \\ \varepsilon &/\left( + } \right) < \varepsilon } \end} \right.$$
(1a)
$$ = \left\}&}/\left( } + CD8} \right) > 1 - \varepsilon } \\ }}}} + CD8}}}&}/\left( } + CD8} \right) \leqslant 1 - \varepsilon } \\ \varepsilon &}}}} + CD8}} < \varepsilon } \end} \right.$$
(1b)
$$ = \left\}& \right) > 1 - \varepsilon } \\ }}}& \right) \leqslant 1 - \varepsilon } \\ \varepsilon & \right) < \varepsilon } \end} \right.$$
(1c)
Here, \(_, _, _, CD8\) and \(CD4\) are the tumor densities of the M1 and M2 macrophages, and regulatory, CD8 and CD4 T cells. These immune subset ratios are a modification of the ones presented by [10]. In particular, we add a new parameter \(\epsilon\), which is a positive, and less than 1, divergence-avoiding parameter. With this definition, we are able to avoid any singularities associated with very small or zero denominators when we consider simple ratios of cell numbers. We then compare the ratios in plausible patients with the same immune subset ratios constructed using the omics data from the iAtlas database. The comparison follows the procedure set by [12]. Similarly to [10], we adopt the ratios for comparison because the iAtlas data (i.e., proportions of immune subsets in leukocytes) do not directly correspond to the unit of cell densities in our model (cells/mL tumor). Thus, we estimate the probability density of the immune subset ratios based on their distributions in iAtlas data and plausible patients. Then, we define the probability of inclusion as the ratio of probability density between iAtlas data and plausible patients. In front of this ratio is a normalization factor \(\beta\), which is obtained via simulated annealing [12]. Using this probability of inclusion, we proceed to select the virtual patients from our plausible patient cohort. A detailed algorithm can be found in the Supplementary information (Fig. S1). Finally, this virtual patient cohort is used to conduct virtual clinical trials. While the iAtlas database contains a plethora of omics data of many kinds, we have selected to use the three immune subset ratios since the involved immune cells are explicitly modeled in our framework.
Fig. 1
(a) Algorithm for virtual patient selection based on pre-treatment immune subset ratios derived from iAtlas omics database. (b) Quantitative Systems Pharmacology model for checkpoint inhibition using Pb-Tx. Natural death/degradation of cellular/molecular components was omitted from the figure. The figure also includes a cartoon showing the Pb-Tx unmasking, cleaving and binding
Overview of the QSP-IO model informed with PbTx dynamicsQSP-IO models are systems biology type frameworks used to predict population and individual responses to immuno-, chemo- and combination therapies. Usually, the systems in question are divided into multiple compartments, each representing a different tissue or organ or their assemblies, inside which a set of explicitly modeled molecular or cellular species can interact or move between compartments. In particular, QSP-IO models are informed with the PKPD relations governing the drug as well as the interactions between the immune system and tumor cells. Each simulation is run for a different virtual patient which is the set of parameters describing the mechanistic relations or properties of each species and compartment.
In this work, we implement the previously proposed QSP-IO model for the evaluation of pacmilimab monotherapy, a PbTx molecule, as the anti-PD-L1 immune checkpoint inhibitor [6]. While we provide a detailed explanation of the theory and implementation in the Supplementary information, here we give a brief overview of the model.
The model used in our work, shown in Fig. 1b, contains 4 distinct compartments: the tumor, tumor-draining lymph node, peripheral, and central compartment. The tumor compartment contains the cancer and stromal cells and is the location where the tumor interacts with the components of the immune system in the TME. The tumor-draining lymph nodes are the location where antigen presentation and T cell maturation occur. The peripheral compartment describes the rest of the body and it is generally used to describe unwanted activity of the therapy. Finally, the central compartment represents blood flow which connects all the compartments.
The model contains the dynamics of several different cell types from the immune system as well as those of the tumor. These dynamics are divided into 11 different modules, each describing different cells or key molecular species interactions. Within these modules, we explicitly model the maturation, activation, depletion, and death of CD8, CD4 and regulatory (Treg) T cells. These cells come into contact with mature antigen presenting cells (APCs) arriving from the TME. These APCs mature due to the release of cytokines in the TME. Additionally, macrophages in the M1 and M2 configurations are also modeled in the TME. The interactions between these different cells are ascribed to the immune synapse compartments. This compartment includes all the different ligands used for immune-regulatory actions, such as PD-L1, PD-1, PD-L2, CTLA4, CD28, TCR, MHC and CD80, in the synapse. Of the 11 modules, 10 were previously implemented by Wang et al. [9] and the additional compartment to explicitly model the out-of-synapse dynamics was introduced by Ippolito et al. [6]. We explicitly include only PD-L1 in the out-of-synapse compartment, since it is the only ligand that interacts with the Pb-Tx in this study. However, we want to emphasize that the same procedure can be applied to the other ligands, i.e. CTLA4 and PD-1 and we still explicitly model the interactions between PD-L1, PD-1, PD-L2, CTLA4, CD28, TCR, MHC and CD80 in the synaptic compartment as these are immune-regulatory.
Pacmilimab is an antibody made into a Pb-Tx by the addition of a mask covering the active sites and kept in proximity thereto by a cleavable substrate linker. The PK model describing the Pb-Tx dynamics has been described previously by Stroh et al. [4, 5]. Briefly, the mask is allowed to stochastically and reversibly shift and reveal the active site. This unmasking rate is governed by an equilibrium constant \(_\), which is the likelihood that the mask will reveal the active site. If this were the only addition to the antibody, then the Pb-Tx would just be a less active form of its parent solely due to the reduced exposure of the active site. However, this reduced exposure is made selective since the key property of the Pb-Tx is that the substrate holding the mask is cleavable by proteases that are active in the TME. These proteases are tumor-associated serine protease matriptase, urokinase plasminogen activator and cysteine protease legumain [1] for pacmilimab; however, it is possible to select other substrates that are digestible by different enzymes [15, 16]. We calibrate the model using previously published model parameters from [10], such as the tumor growth rate [17], with the addition of the Pb-Tx specific parameters from Ippolito et al. [6] calibrated to NSCLC by modifying the \(_\) to represent the tumor protease activity. In particular, we have shifted the median of the patient distribution of \(_\) according to the expressions of the enzymes in [15, 16] and we have maintained the same variation interval described in [6]. All species, chemical reactions, ordinary differential equations governing the processes, and model parameters are provided in the Supplementary information files.
Calibration of the immune subset ratiosIn the previous section we described the virtual patient selection criteria using omics-informed immune cell ratios. Here we will describe the calibration of these ratios and the selectivity of the probability of the inclusion imposed by each immune ratio or constraint.
The probability of inclusion is built based on the log of the immune ratios [10], thus it is subject to both positive and negative divergences, i.e. plus and minus infinity. Compared to previous work [10], the positive divergences are avoided by defining the constraints as immune cell ratios instead of fraction. On the other hand, the negative divergence is avoided by introducing the constant \(\epsilon\) which bounds the maximum and minimum value of the ratio between \(1-\epsilon\) and \(\epsilon\), as shown in Fig. 2a. We proceed evaluating the relative error of the mean, shown in Fig. 2a, defined as \((}_ \left(\right)-}_)/}_\), where \(}_ \left(\right)\) is the median of the iAtlas immune fraction distribution modified by \(\epsilon\) and and \(}_\) is the median of the original iAtlas distribution. The dips in the plot correspond to when \(\epsilon\) is equal to the median of the distribution of the given ratio \(_\) (see Fig. S2 for distribution comparisons).
Once the ratios \(_\) were defined, we analyze how constraining or how low they reduce the acceptance rate of virtual patient selection from the plausible patient cohort. We summarize the results in the table shown in Fig. 2b, where we begin from the same cohorts of proposed patients (and plausible patients) and we then calculate the probability of inclusion and select the virtual patients from the initial set. We report the percentage of initial proposed patients finally accepted as virtual. We noticed that, when applying only a single ratio as the constraint, the \(_\) was the one that allowed for the lowest percent compared to the other 2 ratios (9.7 vs. 14.2 and 12.3 for \(_\) and \(_\) respectively). We assume that this may be due to the property of the model, since CD4 cells include a spectrum of cell types which we may not be representing fully. We note that amongst the CD4 cells we explicitly model both Tregs and T helper cells. Additionally, by applying more constraints simultaneously the final acceptance rate is lower than the acceptance rate of either constraint applied singularly. This observation is reasonable since applying more constraints is expected to make a more stringent selection rate for VPs from plausible patients.
Fig. 2
(a) Definition of the immune ratios \(_\), \(_\) and \(_\). The relative error in the mean distribution of the ratios is shown as a function of \(\epsilon\). (b) Acceptance rate percentage of the proposed patients as virtual patients for each combination of immune-omics constraints
留言 (0)