
記住我
Precise activity of cellular protein networks is essential for the proper function and fitness of cells and organisms. Protein function highly depends on its three-dimensional structure, i.e., its folding state (Yadav et al., 2019). To ensure functionality, biological systems have evolved protein quality control mechanisms comprising molecular chaperones, proteases and regulatory factors, which supervise protein folding and direct protein species which fail to meet cellular quality control criteria for degradation. Despite the existence of highly specialized protein folding surveillance mechanisms, protein misfolding and aggregation are hallmarks of numerous human diseases (Chiti and Dobson, 2006; Chiti and Dobson, 2017). These diseases range from systemic amyloidoses, where amyloid fibrils accumulate throughout the human body, to neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and ALS, where protein aggregates are mainly deposited either extracellularly (AD) or intracellularly (PD, ALS) within the central nervous system (CNS).
ALS is a neurodegenerative disease (Hardiman et al., 2017) characterized by the selective degeneration of both upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord (Tandan and Bradley, 1985; Hardiman et al., 2017; van Es et al., 2017). It is the most common form of motor neuron disease with adult onset and the third most common neurodegenerative disease (Hou et al., 2019). The survival rate is highly variable, with a median rate of about 3–5 years after symptom onset; however, up to 10% of ALS patients survive for more than 10 years (Chio et al., 2009; van Es et al., 2017; Longinetti and Fang, 2019). ALS affects approximately two to four people per 100,000 individuals per year in Caucasian populations and about one person or less per 100,000 individuals annually in Asian and Hispanic populations (Cronin et al., 2007; Al-Chalabi and Hardiman, 2013; Hardiman et al., 2017; Longinetti and Fang, 2019), with the exception of Japan where the incidence rate is closer to the that of Western countries (Doi et al., 2014). Recent studies indicate a trend of rising incidence rates for ALS (Collaborators, 2019; Longinetti and Fang, 2019; Ryan et al., 2019).
ALS has traditionally been classified as either sporadic (sALS), representing approximately 90%–95% of cases, or familial (fALS), typically characterized by Mendelian autosomal dominant inheritance (van Es et al., 2017; Ranganathan et al., 2020; Kirola et al., 2022). The term “sporadic” is applied to cases which are unrelated to family incidence, even when gene mutations associated with or causing ALS are present (Mejzini et al., 2019). Comprehensive analyses from large-scale genome-wide association studies (GWAS) on sALS patients have revealed that the genetic landscape of ALS predominantly comprises rare variants (van Rheenen et al., 2016; van Rheenen et al., 2021). Consequently, the field is progressing toward a more precise genetic classification, emphasizing the identification of risk genes (Cooper-Knock et al., 2021; Brenner and Freischmidt, 2022). Regardless of the subclassification criteria, both forms of ALS are clinically indistinguishable (Couratier et al., 2021), differing only in the age of onset, with fALS occurring about a decade earlier than sALS (Wijesekera and Leigh, 2009).
Familial cases of ALS have facilitated the identification of the involvement of the genetic background in disease pathogenesis (Gros-louis et al., 2006). Currently more than 40 genes have been associated with fALS (van Rheenen et al., 2021; Brenner and Freischmidt, 2022; Suzuki et al., 2023), enabling the generation of animal models carrying ALS-related gene mutations (Bonifacino et al., 2021; Zhu et al., 2023). In vivo modelling of ALS has provided a valuable tool to elucidate the pathogenic mechanisms contributing to disease onset and progression, and allowing for targeted drug development (Hardiman et al., 2017; Bonifacino et al., 2021; Zhu et al., 2023). Importantly, research of ALS animal models has revealed an intertwining network of molecular (e.g., aberrant RNA metabolism, impaired protein homeostasis, oxidative stress, mitochondrial dysfunction, impaired DNA repair and dysregulated vesicle transport) and cellular disruptions (such as hyperexcitability, glial dysfunction and axonopathy) that build up to systemic aberrations, ultimately leading to the disease (Hardiman et al., 2017).
ALS is a complex disease, characterized by significant clinical heterogeneity. This heterogeneity is evident in the variability of the site and age of disease onset, the rate of progression, and the degree of cognitive impairment (Chio et al., 2011; Ferrari et al., 2011; Statland et al., 2015). Despite this clinical variability, a cellular hallmark of ALS is the presence of ubiquitinated skein-like or dense and round cytoplasmic inclusions of certain proteins, such as SOD1, TDP-43, and fused in sarcoma (FUS) in motor neurons (Leigh et al., 1988; Lowe et al., 1988). Approximately 97% of ALS cases exhibit TDP-43-positive inclusions (Mackenzie et al., 2007). Notably, TDP-43 cytoplasmic inclusions are also found in other neurodegenerative disorders including Frontotemporal lobar degeneration (FTLD), combined ALS-FTLD, AD and atypical Parkinsonism (de Boer et al., 2020). TDP-43 inclusions are absent in ALS cases caused by mutations in SOD1 and FUS; in these instances, protein deposits consist of the respective mutated gene products (Neumann et al., 2009; Vance et al., 2009).
Considering that aberrant cytoplasmic inclusions are a ubiquitous finding in ALS patients, numerous research efforts have been directed toward inhibiting the aggregation of ALS-related proteins. In this review, we focus on two extensively studied ALS-related proteins associated with protein misfolding and aggregation, SOD1 and TDP-43. We examine the contribution of their accumulation to disease phenotypes and review the recent progress in identifying inhibitors of their aggregation, which could serve as lead molecules for the development of effective anti-ALS treatments.
Misfolding- and aggregation-mediated toxicity in ALSAggregation-prone proteins in ALSSOD1 plays a major role in regulating redox potential by catalyzing the conversion of the superoxide anion O2− into hydrogen peroxide, which is further processed by other enzymes (Bunton-Stasyshyn et al., 2015; Huai and Zhang, 2019). In its functional form, SOD1 forms a homodimer, with each subunit containing a copper and a zinc ion at its [Cu/Zn] site and an intra-subunit C57-C146 disulfide bond (Valentine et al., 2005; Huai and Zhang, 2019). The discovery of SOD1 mutations causing fALS in 1993 was a milestone for ALS research (Rosen et al., 1993). To date, more than 200 mutations in human SOD1 have been associated with ALS (van der Spek et al., 2019). Mutations in SOD1, lead to classical dominantly inherited ALS, accounting for about 10%–15% of fALS cases and approximately 1.5% of sALS cases (Gros-louis et al., 2006). Although SOD1 misfolding and aggregation are considered hallmarks of SOD1-fALS (Saccon et al., 2013), SOD1-immunoreactive inclusions have also been detected in a significant percentage of sALS cases (Bosco et al., 2010; Forsberg et al., 2019). As the longest-studied ALS-related protein, SOD1 has been associated with a large number of pathophysiological mechanisms causing neurotoxicity, including protein misfolding, excitotoxicity, oxidative stress, impaired axonal transport, inflammation and mitochondrial dysfunction (Ferraiuolo et al., 2011).
Wild-type SOD1 is an unusually stable protein, which remains active even under denaturing conditions (Arnesano et al., 2004). It is able to maintain its disulfide status inside the reducing cytosolic environment and is highly resistant to proteolysis (Arnesano et al., 2004; Valentine et al., 2005). This remarkable stability is regulated by the disulfide bond and the metalation status, which mutually affect each other (Arnesano et al., 2004; Tiwari et al., 2005; Nordlund et al., 2009). Disruption of the disulfide bond or loss of the metal co-factors can lead to pathogenic misfolding. Specific post-translational modifications (PTMs), including phosphorylation, acetylation, and succinylation, play crucial roles in regulating the structure and functions of SOD1, such as ROS scavenging, cytoskeletal organization, and transcriptional activity (Banks and Andersen, 2019), while others like sumoylation and oxidation can lead to misfolding and aggregation (Fei et al., 2006; Xu et al., 2018; Trist et al., 2022).
Interestingly, mutations associated with ALS have been identified across all five exons of human SOD1. These mutations impact the structure of SOD1 in different ways, often leading to varying degrees of misfolding, aggregation and consequent variable toxicities (Brasil et al., 2018). Initially, it was proposed that ALS-causing mutations in SOD1 were linked to a loss of enzymatic function (Rosen et al., 1993; Saccon et al., 2013); however, subsequent studies in SOD1 mice contradicted this notion (Gurney et al., 1994; Reaume et al., 1996). Mutant SOD1 retaining partial or complete enzymic function was found to induce ALS-like phenotypes (Gurney et al., 1994), and Sod1 knockout mice did not develop ALS (Reaume et al., 1996). In contrast, ALS-related SOD1 variants are associated with decreased metal binding, reduced formation of a stabilizing intramolecular disulfide bond, diminished structural stability and an increased tendency to monomerize and aggregate (Ray et al., 2004). Importantly, the aggregation propensity of several SOD1 variants has been linked to life expectancy after the onset of ALS symptoms in both humans and transgenic mouse models (Wang et al., 2008; Prudencio et al., 2009; Pratt et al., 2014; Lang et al., 2015; McAlary et al., 2020).
TDP-43, encoded by the TARDBP gene, was identified in 2006 as a major component of cytoplasmic protein inclusions accompanied by nuclear clearance of the protein, as observed in motor neurons of ALS cases (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2007). TDP-43 pathology is present in nearly all ALS cases except for those caused by mutations in SOD1 or FUS. TARDBP mutations account for approximately 5% of familial and almost 1% of sporadic ALS cases. It is an RNA-binding protein (RBP) involved in the regulation of gene expression at multiple levels by playing an active role in alternative splicing of pre-mRNAs (Bhardwaj et al., 2013; Lukavsky et al., 2013; Kuo et al., 2014), splicing of non-coding RNAs (Tollervey et al., 2011) and mRNA stability (Fukushima et al., 2019), thus affecting diverse cell processes, including mitochondrial homeostasis (Izumikawa et al., 2017), DNA damage response (Konopka et al., 2020; Riancho et al., 2020; Wood et al., 2020) and axonal transport (Fallini et al., 2012; Tripathi et al., 2014). While TDP-43 primarily localizes in the nucleus, upon cellular stress, it translocates to the cytoplasm to form stress granules together with other RBPs and stalled ribosomes (Colombrita et al., 2009). Interestingly, single cell proteomic analysis of somatic motor neurons (MNs) derived from postmortem spinal cords of ALS donors with TDP-43 pathology, not only facilitated the differentiation of disease states in individual MNs but also uncovered significant reduction in the abundance of proteins with critical roles in cell energetics, protein translation, proteostasis, and trafficking mechanisms such as Golgi-lysosome trafficking (Guise et al., 2024). Furthermore, C9orf72 mutations lead to the formation of RNA foci which sequester a range of RBPs including TDP-43, resulting in its aberrant accumulation in the cytoplasm (Lee et al., 2013; Chew et al., 2019).
TDP-43 consists of four domains that mediate distinct activities: (a) a globular N-terminal domain (NTD) with a nuclear localization signal (NLS). The NTD is crucial for the formation of the TDP-43 functional dimer/oligomer (Qin et al., 2014; Mompean et al., 2016; Afroz et al., 2017; Jiang et al., 2017; Mompean et al., 2017) and recruits RNA for splicing (Jiang et al., 2017); (b) two tandem RNA recognition motifs (RRM1 and RRM2) (Lukavsky et al., 2013) with a nuclear export signal (NES) in RRM2 (Winton et al., 2008), although the NES functionality has been questioned (Archbold et al., 2018; Pinarbasi et al., 2018), and (c) a prion-like domain (PrLD) at the C-terminus encompassing two subdomains, a glutamine/asparagine-rich and a glycine-rich region, which is disordered and essential for protein-protein interactions (Buratti, 2020; Jiang et al., 2013). Six cysteine residues are present in TDP-43, with four located in the two RRM domains (Cys173, Cys175, Cys198, and Cys244), while the other two (Cys39 and Cys50) in the N-terminal domain (Valle and Carri, 2017). The oxidation of cysteines within the two RRMs decreases protein solubility and induces the formation of intra- and inter-molecular disulfide linkages (Cohen et al., 2012; Chang et al., 2013). The PrLD mediates TDP-43 intrinsic aggregation propensity (Johnson et al., 2009) and its incorporation into stress granules via its ability to undergo liquid-liquid phase separation (LLPS) (Conicella et al., 2016; Wang et al., 2018). Interestingly, the majority (∼95%) of the ALS-linked TARDBP mutations are localized at the C-terminal domain (Prasad et al., 2019); these TDP-43 variants show increased stress granule formation upon oxidative stress (Liu-Yesucevitz et al., 2010) and higher aggregation propensity (Conicella et al., 2016). Moreover, certain TARDBP-linked mutations enhance cytoplasmic mislocalization of TDP-43 (Barmada et al., 2010; Mutihac et al., 2015; Mitsuzawa et al., 2018). Finally, in addition to cysteine oxidation, also other PTMs such as phosphorylation, acetylation, ubiquitination and the proteolytic processing of TDP-43 that leads to the formation of C-terminal fragments are closely associated with the misfolding and aggregation of the protein (Buratti, 2018; Farina et al., 2021).
Cell culture and animal models have provided compelling evidence indicating a significant involvement of TDP-43 in the initiation and progression of motor neuron degeneration (Bonifacino et al., 2021). Transgenic mice overexpressing TDP-43 with familial fALS mutations, develop aggregates and manifest a full spectrum of ALS-like phenotypes at the molecular, cellular and behavioral levels (Ke et al., 2015; Huang et al., 2020). Similarly, the overexpression of a TDP-43 variant lacking the nuclear localization signal (dNLS), leads to the accumulation of insoluble, phosphorylated cytoplasmic TDP-43 in the brain and spinal cord. This is accompanied by brain atrophy, muscle denervation, significant motor neuron loss, and the development of progressive motor impairment (Walker et al., 2015). Notably, suppression of TDP-43 overexpression leads to a rapid clearance of TDP-43 pathology and rescues motor deficits in inducible mouse models (Ke et al., 2015; Walker et al., 2015). These collective findings strongly suggest that TDP-43 is a key protein implicated in neurodegenerative processes in motor neurons.
The aggregation processThe term protein aggregation describes the transition of a protein from its native biologically functional state to the formation of oligomers and medium- or higher-order aggregates through self-association. Protein aggregation is usually caused by the presence of unfolded or misfolded species of the protein. Misfolded conformations typically expose hydrophobic segments within a hydrophilic- either intracellular or extracellular- environment, which subsequently tend to self-associate into soluble oligomers and eventually to larger insoluble aggregates. Most often, the building block of oligomeric and aggregated species is the protein’s unfolded/misfolded/partially folded monomer. The structure of such aggregates highly depends on the conformation of the monomeric state: disordered amorphous aggregates are usually formed by unfolded or native monomers while well-defined fibrils with cross-β structure (amyloid fibrils) can originate from partially folded monomers (Almeida and Brito, 2020). Amyloid deposits have been extensively implicated in disease pathogenesis, while there are few disorders associated with amorphous aggregates, such as cataracts caused by γD-crystallin disordered deposition (Moreau and King, 2012).
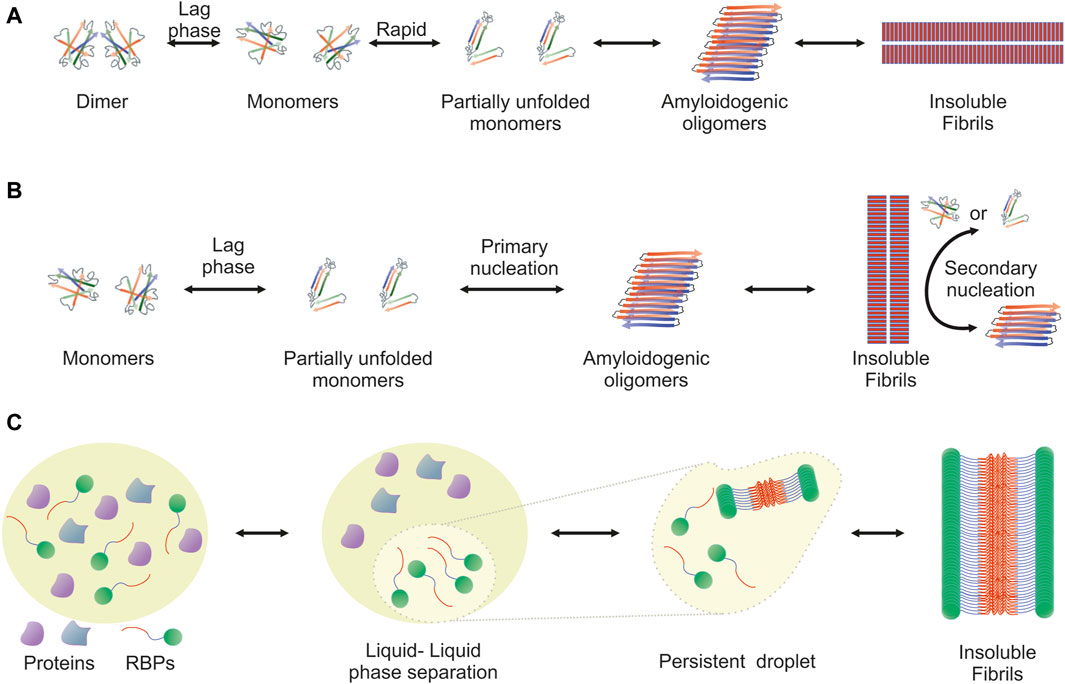
The amyloidogenic pathway has been extensively studied and two distinct mechanisms, the downhill polymerization and the nucleation-growth model, have been described. Both are characterized by sigmoidal kinetics and differ in the factor that determines the rate-limiting step. In each case, the mechanism of aggregation employed depends on the protein. Downhill polymerization (Figure 1A) is typically observed in proteins with oligomeric native conformation, such as transthyretin (TTR) (Hammarstrom et al., 2003; Eisele et al., 2015). The rate-limiting step is the disassembly of the native oligomer into unstable monomers, which then rapidly form aggregates (Lai et al., 1996; Hurshman et al., 2004; Eisele et al., 2015). Importantly, the lag phase is governed by the slow dissociation of native oligomeric state and seeding does not accelerate the aggregation process. The nucleation-growth model (Figure 1B), characteristic of the aggregation of amyloid-β peptide (Knowles et al., 2014), resembles the crystallization process (Teplow, 1998; Scherzinger et al., 1999; Wood et al., 1999; Knowles et al., 2014). The rate-limiting step for amyloid fibril development is the formation of protein oligomers (nuclei or seeds), which subsequently drive the rapid elongation of the fibrils by further monomeric or oligomeric species binding to the nuclei. The slow lag phase can be eliminated by adding pre-formed nuclei.
Figure 1. Visualization of the amyloidogenic pathway featuring three distinct models. Detailed overview of the diverse mechanisms through which amyloid fibrils can form, each highlighting different aspects and stages of the amyloidogenic process. (A) Downhill Polymerization Model: The protein first dissociates into native monomers, which is the rate-limiting step in the formation of fibrils. Subsequently, the monomeric species partially unfold to form the aggregation intermediates. Once such intermediates are formed, the following self-assembly process is a downhill polymerization. (B) Nucleation-Growth Model: Amyloid aggregation is kinetically dependent on nucleation events, which include primary and secondary nucleation. Primary nucleation refers to initial events where protein monomers associate into nucleation “seeds.” This is a rate-limiting, kinetically unfavorable process, but once the nuclei are formed, addition of further monomers is more favorable through an elongation process. Primary nucleation causes elongation into fibrils, which eventually leads to the formation of a critical mass of fibrils capable of catalyzing a secondary nucleation phase in which nucleus formation from monomers is catalyzed by existing aggregates. This creates a positive feedback loop of aggregation, where small and large oligomers are constantly formed. (C) Liquid-Liquid Phase Separation (LLPS) Model: It proposes that amyloidogenic proteins first undergo liquid-liquid phase separation, forming protein-rich droplets. Within these droplets, the concentration of proteins reaches a threshold that facilitates the transition from a disordered liquid state to a more ordered solid state, leading to amyloid fibril formation. This model provides insight into the spatial and temporal regulation of amyloidogenesis within cellular environments.
Another mechanism that leads to the formation of ordered aggregates is mediated by LLPS, typically observed in RBPs (Figure 1C). Approximately 30% of the genes associated with ALS encode RBPs, such as TDP-43, FUS, TATA-box binding protein associated factor 15 (TAF15) and proteins from the heterogeneous nuclear ribonucleoprotein (hnRNP) family, including hnRNPA1 and hnRNPA2B1 (Wroe et al., 2008). These proteins form cytoplasmic inclusions in motor neurons of ALS patients and their aggregation propensity is linked to the PrLDs they harbor (Molliex et al., 2015; Patel et al., 2015). PrLDs are a subset of intrinsic disordered regions (IDRs), characterized by low sequence complexity, comprising only a subset of the 20 amino acids; in particular, PrLDs are enriched in uncharged polar amino acids and glycine (Alberti et al., 2009). Intriguingly, about 30% of the 240 human proteins containing predicted PrLDs are involved in RNA processing (March et al., 2016; Verdile et al., 2019). LLPS describes liquid-liquid de-mixing: a high local concentration of IDRs promotes LLPS through transient and weak interactions among the IDRs of RBPs. Importantly, association of RBPs into “droplets” which are separated from the nucleoplasm or the cytoplasm is dynamic and transient. However, persistent association of RBPs with the separated liquid state results in the formation of stable hydrogels and eventually fibrils (Kato et al., 2012; Lin Y. et al., 2015; Guo and Shorter, 2015; Molliex et al., 2015; Murakami et al., 2015; Patel et al., 2015; Kato and McKnight, 2017; Shin and Brangwynne, 2017).
Until recently, there was intense debate over whether the protein aggregates detected in ALS patients are typical amyloids, primarily due to controversial findings. Initially, reports indicated that the TDP-43 positive filamentous aggregates found in ALS-affected motor neurons (Hasegawa et al., 2008; Lin and Dickson, 2008) did not stain with the amyloid-specific dyes Congo red and Thioflavin S (ThS) (Kerman et al., 2010). This finding was further supported by in vitro experiments, where recombinant TDP-43 formed Thioflavin T (ThT)-negative granulo-filamentous aggregates similar to those observed in ALS patients (Johnson et al., 2009) or amorphous aggregates (Capitini et al., 2014). On the other hand, it has been shown that peptide fragments from the C-terminal domain and certain TDP-43 variants form ThT-positive aggregates in vitro, acquiring a β-sheet-enriched structure (Chen et al., 2010; Guo et al., 2011; Zhu et al., 2014; Jiang et al., 2016). These conflicting findings can now be explained based on the structure of TDP-43 aggregates from ALS with FTLD (Arseni et al., 2022), revealing the formation of filaments which are structurally distinct from cross-β amyloid fibrils and a novel fold that bears no similarity to TDP-43 filaments formed in vitro (Cao et al., 2019; Li et al., 2021).
The Falcon group employed cryogenic electron microscopy to determine the structures of aggregated TDP-43 in the frontal and motor cortices of an individual who succumbed to ALS with FTLD, as well as in the frontal cortex of another individual with the same diagnosis (Arseni et al., 2022). Remarkably, they identified an identical amyloid-like filament structure consisting of a single protofilament in both brain regions and individuals. The ordered filament core, spanning residues 282 to 360 in the TDP-43 low-complexity domain, adopts a unique double-spiral-shaped fold. The filament structure lacks the typical β-sheet stacking associated with cross-β amyloid formation due to an abundance of glycine and neutral polar residues, facilitating numerous turns and restricting β-strand length. The uneven distribution of residues results in structurally and chemically distinct surfaces, suggesting potential ligand binding sites on external densities.
This study also revealed that the brain-derived TDP-43 filaments are structurally distinct compared to filaments assembled in vitro from its LCD under mildly acidic conditions (pH 4), or related fragments (Li et al., 2021). These differences include opposite chirality and variations in both protein fold and secondary structure. For the LCD, the structure revealed single protofilament fibrils featuring a large core comprised of 139 residues out of the 148 present in the LCD, tightly packed together. The C-terminal segment of this core exhibits mainly planar characteristics and is marked by a small proportion of hydrophobic amino acids. In contrast, the N-terminal region includes numerous hydrophobic residues and adopts a non-planar backbone conformation, leading to the rugged surfaces observed at the ends of the fibrils.
Cryo-electron microscopy was also employed to elucidate the structures of two segments identified as the pathogenic cores in human TDP-43 aggregation (Cao et al., 2019). The first segment, SegA (residues 311–360), exhibits three polymorphs, each characterized by a dagger-shaped fold, spanning residues 312–346. Tight hydrophobic interactions are formed by hydrophobic residues ranging from Phe313 to Ala341 whereas the dagger tip is created by a sharp 160° kink at Gln327. The variations among the three SegA polymorphs primarily arise from differences in protofilament numbers and symmetry. In contrast to the polymorphic nature of SegA, the second segment, SegB A315E (residues 286–331, containing the ALS hereditary mutation A315E), forms fibrils with a consistent morphology. These fibrils adopt an R-shaped fold spanning residues 288–319, which, overall, displays a more pronounced kink compared to the dagger-shaped fold, likely attributable to the higher prevalence of glycine residues. Each fibril is composed of four protofilaments, and notably, all four R-folds are characterized by the presence of a salt-bridge between Arg293 and Glu315, facilitated by the pathogenic A315E mutation. This salt-bridge may impact the kinetics of fibril growth and nucleation, providing a mechanistic explanation for A315E’s propensity to promote TDP-43 aggregation through electrostatic attraction with Arg293.
Along the same lines, SOD1-positive inclusions from ALS cases display a fibrillary morphology (Kato et al., 2000) but do not react with ThS (Kerman et al., 2010). In contrast, SOD1 cytoplasmic aggregates from a transgenic SOD1-fALS mouse model could be stained with ThS (Furukawa et al., 2008). Importantly, the aggregation kinetics of SOD1 are typically monitored by ThT staining (Rakhit et al., 2002; Furukawa et al., 2008; Iwakawa et al., 2017; Shvil et al., 2018) and pre-formed SOD1 aggregates exhibit seeding activity both in vitro and intracellularly (Furukawa et al., 2008; Furukawa et al., 2013). Moreover, ALS-related SOD1 mutant proteins crystallize in forms comprising higher-order assemblies of aligned β-sheets (Elam et al., 2003). On the other hand, in vitro monitoring of SOD1 aggregation has demonstrated that a competition between amorphous and amyloid aggregation occurs (Abdolvahabi et al., 2016), proposing a possible factor for the wide variability in kinetic results among publications (Crown et al., 2019).
In a recent study, cytotoxic amyloid fibrils were generated from full-length human apo-SOD1 under reducing conditions, and their atomic structure was elucidated using cryo-EM (Wang LQ. et al., 2022). The SOD1 fibril is composed of a singular protofilament featuring a left-handed helix. Within the fibril core, a serpentine fold is formed, incorporating six β-strands of the N-terminal segment (residues 3–55) and seven β-strands of the C-terminal segment (residues 86–153), with an unstructured region in between. This novel amyloid fibril structure displays a very compact fold, and the connection of the two segments is stabilized by three pairs of salt bridges, effectively “zipping up” the structure.
Characteristics of oligomeric speciesA multitude of pathogenic mechanisms have been described for ALS, including protein misfolding and aggregation, glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, declined autophagy, neuroinflammation and DNA damage. However, the drive behind ALS onset and progression, as well as the degree to which each mechanism contributes to disease phenotypes and heterogeneity, remains unclear. Current evidence supports that protein misfolding and aggregation exert toxic effects on cellular fitness, eventually compromising organismal health. Regarding the toxic agent in ALS and other conformational neurodegenerative diseases, the debate continuous on whether soluble misfolded species (monomeric or oligomeric) or insoluble aggregates confer toxicity (Gelpi and Colom-Cadena, 2019).
Recent work has highlighted SOD1 oligomers, particularly the trimeric variants, as the neurotoxic entities (Proctor et al., 2016). The study revealed that SOD1 mutants promoting trimerization led to increased neuronal cell death and established a direct association between misfolded oligomers and neuron death by linking cytotoxicity to trimer stability. Additional studies showed that large aggregates are non-toxic and actually play a neuroprotective role (Zhu et al., 2018; Gill et al., 2019). Interestingly, SOD1 mutants designed to promote the formation of large aggregates and destabilize toxic trimers did not impact cell viability (Zhu et al., 2018). The mechanism leading to the formation of non-native trimers remains unclear, however, in order to occur, SOD1 must undergo dissociation into monomers (Khare et al., 2004). External factors, such as oxidative stress (Wilcox et al., 2009) or exposure to the toxin β-methylamino-L-alanine (BMMA) (Proctor et al., 2019), may play a role in the transition of SOD1 dimers into monomers. At present, there are two hypotheses regarding trimer formation (Choi and Dokholyan, 2021). The first suggests that trimers occur on the pathway from monomers to native dimers (dissociating into monomers), then to trimers and finally to larger aggregates. The second posits that trimer formation is an off-pathway phenomenon, meaning it does not lead to the generation of large aggregates, and current experimental evidence favors this scenario (Hnath and Dokholyan, 2022). Indicatively, the abundance of soluble misfolded SOD1 subfractions, in the range from monomeric to trimeric in size, has been associated with a reduced lifespan in SOD1-ALS mouse models (Zetterstrom et al., 2007).
Despite the association of TDP-43 aggregation with disease (Neumann et al., 2006; Hasegawa et al., 2008), several studies have highlighted the functional oligomerization of TDP-43 in physiological contexts. The NTD residues 1–105 and 1–265 induce the formation of homo-oligomeric species in a concentration-dependent manner, initiated by the formation of intermolecular disulfide bonds ultimately leading to its tetramerization (Chang et al., 2012; Jiang et al., 2017). Oligomerization of TDP-43 is crucial for its nuclear RNA splicing activity and, interestingly, serves to prevent the aggregation of the C-terminal TDP-43 within the nucleus of neuronal cells (Afroz et al., 2017; Jiang et al., 2017). The crystal structure of a purified recombinant human TDP-43 fragment (residues 1–80) was determined at a resolution of 2.1 Å, revealing a unique pattern of head-to-tail interactions between monomers, resulting in the formation of solenoid-like polymers (Afroz et al., 2017). In contrast to physiological oligomers, pathological TDP-43 oligomers exhibit distinct characteristics. Detectable in the early stages of aggregation, their formation is accelerated in the presence of disease-associated TDP-43 mutations (Johnson et al., 2009; French et al., 2019). Using a polyclonal TDP-43 oligomer-specific antibody (Fang et al., 2014), pathological TDP-43 oligomers were identified in FTLD patients (Kao et al., 2015).
The TDP-43 oligomers manifest as a diverse spectrum of molecules, ranging from low-molecular-weight species like dimers, trimers, and tetramers, to a variety of high-molecular-weight species (Johnson et al., 2009; Guo et al., 2011; Choksi et al., 2014; Fang et al., 2014; French et al., 2019). Described as heterogeneous structures at the intermediate stage of aggregate formation, the oligomerization process may involve cysteine oxidation, particularly in the initial stages (Bozzo et al., 2016). Extracellular exposure to TDP-43 oligomers demonstrated cytotoxicity in neuroblastome cells (Choksi et al., 2014), and intrahippocampal injection of these oligomers caused damage to hippocampal neurons in wild-type mice (Fang et al., 2014), further substantiating their neurotoxicity. Interestingly, in an animal model expressing the ALS-related TDP-43 (M377V) variant at nearly endogenous levels, there was complete absence of mature insoluble aggregates (Gordon et al., 2019). However, these mice progressively developed motor function deficits concomitant with the loss of neuromuscular junction integrity, thus recapitulating ALS phenotypes (Gordon et al., 2019). Taken together, current evidence is not sufficient to argue towards one or the other direction, though it seems possible that both oligomeric soluble species and insoluble aggregates contribute differentially to ALS pathogenesis (Arnold et al., 2013; Gordon et al., 2019).
The ability to isolate, cultivate, and reprogram cells obtained from patients, such as fibroblasts, into central nervous system (CNS) cells, has introduced a novel approach that complements traditional preclinical in vitro and in vivo models (Ferraiuolo and Maragakis, 2021; Du et al., 2023). There are two primary approaches to this method. Several laboratories have investigated the use of induced pluripotent stem cells (iPSCs), focusing on generating motor neurons from patients with mutations in TARDBP, FUS, SOD1, and C9orf72, or those with sporadic disease (Ferraiuolo and Maragakis, 2021). For instance, motor neurons derived from ALS patients with mutated TDP-43, developed cytosolic aggregates resembling those observed in postmortem tissues and displayed shorter neurites (Egawa et al., 2012). Importantly, the accumulation of insoluble protein inclusions has been observed in numerous studies using iPSC-derived motor neurons (Dimos et al., 2008; Burkhardt et al., 2013; Chen et al., 2014; Seminary et al., 2018), although neuronal cell death is not observed, supporting the non-cell-autonomous hypothesis of ALS neurodegeneration (Ilieva et al., 2009; Wachter et al., 2015).
Limiting the screening approach to monocultured motor neurons restricts the influence of non-neuronal cells on observed phenotypes. In ALS, neighboring cells like astrocytes and microglia significantly impact neurodegenerative phenotypes and neuronal survival (Boillee et al., 2006). Additionally, iPSC technology often results in the loss of the cellular aging signature (Lapasset et al., 2011; Patterson et al., 2012). An alternative method involves trans-differentiating patient fibroblasts into neural progenitors, which can then be further differentiated into neurons or astrocytes (Meyer et al., 2014; Mertens et al., 2016; Tang et al., 2017). These astrocytes exhibit expected toxicity toward co-cultured motor neurons and seem to maintain the associated aging phenotype (Gatto et al., 2021). Interestingly, a study using iPSC-derived motor neurons and astrocytes revealed that recombinant TDP-43 oligomers, can induce neuronal toxicity, leading to increased caspase-3 activity and to apoptosis of neurons but not astrocytes (Smethurst et al., 2020).
Mechanisms of aggregation-mediated toxicityConcerning the mechanisms underlying aggregation-mediated toxicity in ALS and other protein misfolding diseases, two major models have been proposed: the loss-of-function hypothesis and the gain-of-toxic activity hypothesis. While both terms are borrowed from genetics, mutations are not always relevant within the context of misfolding and aggregation. In the majority of ALS cases, misfolded species originate from a wild-type protein devoid of inherited mutations. Thus, in this section we focus on how the aggregation process itself, rather than the genetic background (as reviewed in Kim et al., 2020), can lead to phenotypes resembling loss and gain of activity. Importantly, these mechanisms are not mutually exclusive; in fact, some alterations observed in ALS-related cellular physiology can be better explained by a combination of loss and gain of activity.
The loss-of-function hypothesisIn genetics, the term “loss-of-function” is used to describe a mutation that partially or completely disrupts the activity of a gene product. Accordingly, during the aggregation process, sequestration and often mislocalization of a protein within aggregates, reduce its abundance, thereby leading to impaired activity. Indicatively, most ALS-affected motor neurons exhibit wild-type TDP-43 cytoplasmic inclusions concomitant with TDP-43 clearance from the nucleus, indicating that a loss of TDP-43 splicing activity could contribute to disease phenotypes (Polymenidou et al., 2011; Tollervey et al., 2011). In support of this, Tardbp knockout mice display early embryonic lethality (Kraemer et al., 2010); moreover, TDP-43 is essential for normal motor neuron function in flies (Feiguin et al., 2009) and mice (Yang et al., 2014), as evidenced by motor dysfunction and deficits in the neuromuscular junction after TDP-43 depletion. Conditional Tardbp knockout mice, when crossed with various tissue-specific Cre lines, exhibit diverse cellular and behavioral phenotypes, ranging from electrophysiological abnormalities to deficits in motor movement (Iguchi et al., 2013; Wu et al., 2019). TDP-43 has been demonstrated to interact with RNA transcripts of over 6,000 genes in mice. Accordingly, the reduction of TDP-43 levels in adult mouse brains using antisense oligonucleotides (ASOs) led to the differential regulation of approximately 600 genes, mostly involved in synaptic activity and neuronal development, and to the alteration of 1,000 splicing events (Polymenidou et al., 2011).
Loss of function, at least partially, can also be attributed to SOD1-linked ALS. Although Sod1 knockout mice (Sod1−/−) develop normally and do not display evident ALS-like phenotypes (Reaume et al., 1996), they do exhibit- together with other pathologies-progressive motor neuropathy characterized by peripheral motor axon degeneration (Flood et al., 1999; Fischer et al., 2011; Larkin et al., 2011; Fischer et al., 2012; Shi et al., 2014; Ivannikov and Van Remmen, 2015). Furthermore, recent human genetics studies indicate that SOD1 homozygous loss-of-function can lead to debilitating phenotypes, including progressive loss of motor abilities, tetraspasticity, and hyperreflexia, but not ALS. In contrast, heterozygous carriers remained unaffected (Andersen et al., 2019; Park et al., 2019).
The gain-of-toxic activity hypothesisThe accumulation of insoluble proteins is a prominent feature in ALS pathology, suggesting an interconnection between protein aggregation and disease development. The hypothesis posits that protein aggregates may initiate the disease process by acquiring toxic properties, which are mainly exerted through the sequestration of crucial components of physiological processes, such as RNA metabolism, DNA damage response, synaptic signaling and axonal trafficking, thereby disrupting these cellular functions and contributing to disease progression (Ilieva et al., 2009). For instance, TDP-43 aggregates have been found to contain proteins like the RNA-processing factor Matrin 3 (MATR3) (Tada et al., 2018), the ubiquitin-proteasome system factor Ubiquilin 2 (UBQLN2) (Deng et al., 2011; Williams et al., 2012), the Golgi complex and membrane trafficking factor Optineurin (OPN) (Hortobagyi et al., 2011) and the Rho guanine nucleotide exchange factor 28 (RGNEF) (Keller et al., 2012) involved in synapse formation and dendritic morphogenesis. Other potential mechanisms for the induction of cell toxicity by protein aggregation in ALS include disruption of the proteostasis network, leading to impairment of crucial protein degradation pathways like the ubiquitin proteasome system and autophagy (Medinas et al., 2017; Ramesh and Pandey, 2017; Montibeller et al., 2020). Compelling evidence also connects aggregated SOD1 and TDP-43 to mitochondrial dysfunction and degeneration (Jankovic et al., 2021). TDP-43 aggregates sequester specific microRNAs and mitochondrial proteins encoded by the nuclear genome, with dysregulation of their expression levels leading to mitochondrial dysfunction and oxidative stress (Zuo et al., 2021). Aggregates of mutant SOD1 in the intermembrane space of mitochondria diminish the activity of the electron transport chain (ETC) complexes in rats (Pickles et al., 2013) and affect mitophagy by disabling recruitment of autophagy receptors on damaged mitochondria in N2a cells (Tak et al., 2020).
Alternatively, aberrant protein aggregates can induce a chronic inflammatory response in the brain contributing to disease progression (Appel et al., 2021). In a recent study, Yu et al. demonstrated that TDP-43 induces inflammation in ALS by initiating the release of mitochondrial DNA into the cytoplasm. This, in turn, activates the cytoplasmic DNA-sensing cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway (Yu et al., 2020). Separately, the nuclear factor-kappa β (NF-κB) protein has been reported as a master regulator of inflammation in ALS (Haidet-Phillips et al., 2011); in mutant SOD1 mice, NF-κB signaling becomes activated within glia as the disease progresses (Frakes et al., 2014). In support of these findings, extensive astrocytosis (Kushner et al., 1991; Nagy et al., 1994; Reischauer et al., 2018; Schiffer et al., 1996), microglial activation (Henkel et al., 2004; Turner et al., 2004; Zurcher et al., 2015), as well as increased levels of inflammatory cytokines (Moreau et al., 2005; Liu et al., 2015; Lu et al., 2016) and elevated levels of chitotriosidase (Chit-1) and chitinase-3-like protein 1 (CHI3L1) in cerebrospinal fluid (CSF) correlated to disease progression (Vu et al., 2020), have been detected in ALS patients.
Therapeutic targeting of misfolding and aggregationAmong neurodegenerative diseases, ALS stands out as one of the few for which disease-modifying therapies have gained approval. The current standard of care for ALS patients involves multidisciplinary symptom management, both pharmacological and non-pharmacological, such as nutritional and respiratory support (Hardiman et al., 2017; Mejzini et al., 2019). Clinical trials in ALS have assessed over 60 compounds (Petrov et al., 2017), each with distinct mechanisms of action; however, only four- riluzole, edaravone, AMX0035 and tofersen- have been granted regulatory clearance for clinical use. Riluzole, the first FDA-approved ALS therapy in 1995 (Lacomblez et al., 1996a; Lacomblez et al., 1996b), is not a cure for ALS. Instead, it is a neuroprotective drug that decreases glutamate release into the synaptic cleft by blocking voltage-gated sodium channels on presynaptic neurons, thereby mitigating excitotoxicity (Wang et al., 2004; Bissaro and Moro, 2019). While Riluzole does not modify the course of the disease (Fang et al., 2018), it prolongs the patient’s survival from 6 to 19 months (Andrews et al., 2020). Edaravone is a potent anti-oxidant and a free-radical scavenger, preventing oxidative stress-induced motor neuron death (Jami et al., 2015; Soejima-Kusunoki et al., 2022). A randomized controlled trial conducted in Japan demonstrated the effectiveness of Edaravone in slowing the rate of motor function deterioration, particularly in selected patients with early disease onset and rapid progression (Writing Group and Edaravone MCI-186 ALS 19 Study Group, 2017). Its impact on survival is yet to be determined. Apart from Japan and other Asian countries, its use has been approved by the FDA and Health Canada, but not yet from the EMA. A recently developed oral formulation in the United States is expected to replace the intravenous version (Pattee et al., 2023). AMX0035 was approved in 2022 for the treatment of ALS in the United States and Canada, and it is currently being evaluated in the PHOENIX phase III clinical trial (Nikitin et al., 2023). It is a coformulation of sodium phenylbutyrate, a chemical chaperone that improves endoplasmic reticulum (ER) folding capacity by upregulating heat-shock proteins (Suaud et al., 2011), and taurursodiol (tauroursodeoxycholic acid), an inhibitor of mitochondrial-associated apoptosis (Rodrigues et al., 2003). This therapeutic approach is designed to address various ALS-associated pathophysiological mechanisms such as mitochondrial dysfunction and ER stress that ultimately lead to motor neuron injury and cell death.
Tofersen is an antisense oligonucleotide (ASO) that obtained its initial approval in the United States on April 25, 2023, and in Europe on February 22, 2024, for the treatment of ALS in patients with a confirmed SOD1 mutation. It has been designed to mediate RNase H–dependent degradation of SOD1 mRNA to reduce the synthesis of SOD1 protein (McCampbell et al., 2018; Rinaldi and Wood, 2018). The preclinical rationale supporting the ASO knockdown of SOD1 was very robust, stemming from several years of systematic analysis showing increased survival and enhanced motor performance in SOD1 (G93A) rats and mice and decreased SOD1 protein levels in nonhuman primates (Iannitti et al., 2018; McCampbell et al., 2018). Its intrathecal administration in clinical trials demonstrated a deceleration in the decline of participants with rapidly progressing disease and apparent clinical stabilization in those with slower progressing disease (Miller et al., 2022). During the intervention period, concentrations of phosphorylated neurofilament heavy and neurofilament light (NFL) in plasma and CSF, biomarkers indicative of axonal injury and neurodegeneration, were found to be decreased (Miller et al., 2022). Tofersen is currently being evaluated in the phase III ATLAS study for its ability to delay the clinical onset of ALS in pre-symptomatic individuals with a confirmed SOD1 mutation (Benatar et al., 2022).
The complex nature of ALS genetics, coupled with the disease’s high heterogeneity, presents a great challenge in developing treatment strategies that universally benefit every patient. Consequently, there has been a shift in focus towards identifying converging paths and common pathologies that contribute to motor neuron vulnerability and degeneration in ALS. Among these factors, protein aggregation emerges as a prevalent underlying cause not only in ALS but also in various other neurodegenerative diseases. Despite the diversity of mutations in different genes, leading to potential variations in the aggregated proteins, the shared problem of protein aggregation spans a wide spectrum of patients, encompassing familial ALS (fALS), sporadic ALS (sALS), and ALS with frontotemporal dementia (ALS/FTLD) (Blokhuis et al., 2013). As a result, the pursuit of therapies targeting protein aggregation holds significant promise and represents a crucial step forward in addressing ALS and related disorders (Elliott et al., 2020).
Several approaches have been applied to reduce the levels of toxic variants; these include proteolysis targeting chimeras (PROTACs) inducing protein degradation (Tseng et al., 2023), antibodies (Maier et al., 2018; Pozzi et al., 2019; Afroz et al., 2023; Audrain et al., 2023; Bakavayev et al., 2023), vaccines (Zhao et al., 2019), antisense oligonucleotides (Iannitti et al., 2018; Mead et al., 2023), RNA interference (RNAi) with small RNAs (shRNA and miRNA) (Bravo-Hernandez et al., 2020; Mueller et al., 2020), and CRISPR/Cas9 gene editing (Duan et al., 2020). Another way of reducing the accumulation of toxic aggregates is to restore dysfunctional proteostasis by upregulating chaperones (Kalmar and Greensmith, 2017; Kinger et al., 2023), inducing autophagy (Chen et al., 2020) and activating the proteasome (Webster et al., 2017). Currently, many compounds targeting proteostasis, such as trehalose (Seelos Therapeutics) and Trametinib (GENUV) are being explored in clinical trials. Moreover, PMN-267 (ProMIS Neurosciences), an antibody that targets the formation of misfolded TDP-43, is under preclinical development (Mead et al., 2023). Despite the success of Tofersen, a similar strategy utilizing intrathecally delivered ASOs for C9orf72-associated ALS did not show clinical benefits, leading to the termination of the clinical trial in 2022 (Mead et al., 2023).
In this review, we focus on molecules that directly target the misfolded species (monomers, oligomers or mature aggregates) to (a) stabilize the native protein conformation, (b) restore the native conformation of misfolded species and (c) inhibit protein oligomerization. Such molecules fall into two categories depending on the specificity to bind to their target, namely, chemical and pharmacological chaperones.
The term “chaperone” is borrowed from the name of a class of cellular proteins that assist polypeptide chains in acquiring their native, functionally active, conformation. They participate in (a) the folding of nascent polypeptides (de novo folding) or the refolding of stress-mediated misfolding, (b) the disaggregation of protein aggregates and (c) proteostatic pathways that direct denatured and misfolded/unfolded proteins for degradation (Wentink and Rosenzweig, 2023). However, pharmacological chaperones, unlike proteins, are low molecular weight chemical molecules that exert their action by specifically binding to their target proteins. They typically stabilize an already folded or partially folded protein, protect it from thermal denaturation, prevent proteolytic degradation and also inhibit aggregation, restoring proper steady-state levels (Convertino et al., 2016; Bose and Cho, 2017). Pharmacological chaperones have successfully been used experimentally in vitro and in vivo (Leidenheimer and Ryder, 2014; Hou et al., 2018), and they have entered clinical trials, to restore the function of specific misfolded proteins (Liguori et al., 2020; Tran et al., 2020). For example, Tafamidis has been approved for the treatment of transthyretin amyloidosis (ATTR) (Bulawa et al., 2012; Maurer et al., 2018) and Migalastat has been licensed for the treatment of Fabry disease (FD) in patients carrying specific lysosomal enzyme α-galactosidase A (GLA) variants (Germain et al., 2016).
Chemical chaperones are also low-molecular-weight compounds that lack specificity since they can bind to and stabilize virtually any protein without having a designated binding site (Cortez and Sim, 2014). They can be divided into two groups: osmolytes and hydrophobic compounds. Osmolytes, such as glycerol, produce a hydrophobic environment around proteins by removing water molecules, thereby increasing the free energy of the unfolded state and eventually shifting the equilibrium towards the folded state (Wang and Bolen, 1997; Street et al., 2006). Hydrophobic chaperones facilitate proper folding by directly interacting with the exposed hydrophobic regions of unfolded proteins, resembling the action of molecular chaperones (Kuzuhara et al., 2008). For example, the green tea catechin, (−)-epigallocatechin gallate (EGCG), seems to exert its general anti-amyloidogenic properties by directly binding to unfolded/misfolded proteins (Debnath et al., 2016). As mentioned above, the best-known example of chemical chaperone for ALS treatment is sodium phenylbutyrate (Suaud et al., 2011).
Discovering inhibitors of protein misfolding and aggregationDeveloping a new drug is a complex and challenging process that yields significant benefits for both society and the scientific community. High-throughput screening (HTS) is crucial in the early stages of small-molecule drug development, particularly when insufficient information limits structure-based approaches (Blay et al., 2020). For targets such as oligomeric species and aggregates, which lack well-defined structures and exist in dynamically varying mixtures, HTS requires innovative strategies to discover effective treatments. The utilized methods must be biologically relevant, sensitive, robust, and cost-effective, targeting diseases of significant relevance. HTS integrates biochemical and cell-based assays with computational strategies to identify potential drug candidates. In silico screening employs computational methods to predict interactions between molecules and targets, a critical step given the extensive chemical space. Unlike traditional HTS, which assesses thousands of compounds, virtual screening has the capacity to evaluate billions (Bohacek et al., 1996; Gorgulla et al., 2022), showcasing the expansive scope of modern drug discovery efforts.
In 2005, Ray et al. employed an in silico screening strategy of approximately 1.5 million compounds from commercial libraries (Ray et al., 2005). The objective was to identify molecules capable of stabilizing the SOD1 (A4V) dimer. The screening process yielded 100 hits, and subsequent mutagenesis studies using various in vitro aggregation assay protocols helped narrow down the selection to the 15 most effective compounds. Notably, three of the top five compounds shared pyrimidine-like structures. However, subsequent co-crystal structures of SOD1 with various small molecules and pyrimidine-like compounds challenged the initial assumption that the observed protection was solely due to binding to the hydrophobic cavity formed at the dimer interface. Instead, these structures suggested that the ligands interacted with Trp32, offering new insights into the mechanism of action (Antonyuk et al., 2010; Wright et al., 2013).
Nowak et al. developed (Nowak et al., 2011) an algorithm for the in silico screening of an extensive library comprising 2.2 million small molecules sourced from 11 commercially available databases. The primary objective was to discern compounds exhibiting selective binding affinity for mutant SOD1 over plasma proteins. Their investigation revealed a substantial subset of compounds displaying robust binding capabilities to SOD1, particularly within a hydrophobic cavity encompassing Val7–Gly147–Val148 at the dimerization interface. Notably, in vitro experimentation demonstrated the pronounced inhibitory effects of both isoproterenol and 5-fluorouridine on the aggregation process of mutant SOD1.
In another computational screen involving 4,400 drugs and compounds, three flavonoids, quercitrin, quercetin-3-β-D-glucoside (Q3BDG) and EGCG, were predicted to bind around the dimer interface and stabilize SOD1, potentially inhibiting its aggregation, predictions which were confirmed experimentally (Ip et al., 2017). Interestingly, quercitrin and Q3BDG outperformed EGCG both in silico and in vitro. The tripeptide CGH (Srinivasan and Rajasekaran, 2019), hesperidin and 2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside retrieved from the traditional Chinese medicine database (Huang et al., 2014), and the natural polyphenols, kaempferol, and kaempferide (Srinivasan and Rajasekaran, 2018) have all been predicted to bind SOD1 and inhibit its aggregation, necessitating experimental verification.
Biochemical screensThis type of screens involves the use of a purified target protein of interest to assess the aggregation-inhibitory activity of test compounds. To quantify the outcomes of these assays, various optical methods such as absorbance, fluorescence, or luminescence are employed as readouts (Fang, 2012). For example, a screening assay of 640 FDA-approved drugs, aiming to evaluate their impact on the abnormal oligomerization of SOD1 (G37R) has been conducted in vitro. The primary objective was to identify molecules capable of inhibiting the formation of insoluble disulfide-linked oligomers. Among the extensive drug repertoire, six compounds - simvastatin, lovastatin, mevastatin, miltefosine, alfacalcidol and calcitriol-exhibited remarkable efficacy in almost completely suppressing the increase in solution turbidity (Anzai et al., 2016).
Cellular screensCell-based assays play a crucial role at every stage of the drug discovery process, serving various purposes from target identification and validation to lead optimization and sa
留言 (0)