

記住我









Highly pathogenic coronaviruses cause severe respiratory diseases, extra-pulmonary damages, and even death in humans. Over the past two decades, there have been two pandemic outbreaks caused by coronaviruses: severe acute respiratory syndrome (SARS) in 2002 caused by SARS-related coronavirus (SARS-CoV), and Middle East respiratory syndrome (MERS) in 2012 caused by MERS-related coronavirus (MERS-CoV) (Cui et al., 2019). Coronavirus disease 2019 (COVID-19), the ongoing third coronavirus pandemic caused by SARS-CoV-2, has resulted in unprecedented casualties and socioeconomic burden. COVID-19 can manifest in a range of severity, from mild/moderate clinical symptoms to severe or life-threatening diseases (Chen et al., 2020b). The COVID-19 pandemic poses a significant threat not only to elderly individuals with pre-existing health conditions, but also to healthy adults (Gates, 2020). As of March 2024, it has infected over 770 million people worldwide, resulting in approximately seven million fatalities (https://covid19.who.int).
SARS-CoV-2, like all viruses, relies on cellular host factors and pathways to complete its replication cycle successfully. SARS-CoV-2 enters host cells through interactions with cell-surface receptors and proteases. Once inside, the virus engages with intracellular proteins to exploit host mechanisms that facilitate viral replication and evasion of immune responses (Zhou et al., 2023). In this process, SARS-CoV-2 could inevitably induce cellular destruction, disrupt the host immune response, perturb epigenetic regulation, and impair metabolic homeostasis, contributing to the pathogenesis and progression of diseases. Therefore, a better understanding of virus-host interactions is crucial for comprehending SARS-CoV-2 pathogenesis, which would ultimately aid in the identification of novel therapeutic targets and strategies for COVID-19 and related diseases.
The growing number of individuals recovering from COVID-19 has resulted in a heightened focus on long-term effects of this pandemic. Long-COVID refers to a collection of prolonged symptoms that emerge during or after a confirmed or suspected case of COVID-19 (Monje and Iwasaki, 2022). These symptoms encompass various organ systems and commonly include fatigue, breathlessness, headaches, nausea, vomiting, anxiety, depression, skin rash, joint pain, and palpitations (Raman et al., 2022). Moreover, symptoms are merely superficial manifestations. SARS-CoV-2 infection can have lasting and far-reaching effects on the physiological mechanisms and functions of various organs and tissues, not only during the post-acute phase but also persistently in the long term. While long-term organ damage resulting from acute-phase infection is likely responsible for these lasting effects, there may be specific mechanisms following the initial illness that contribute to subsequent pathological changes (Castanares-Zapatero et al., 2022). Therefore, the investigation of host-virus interactions is significant for the prevention and treatment of long-term effects of COVID-19.
This review aims to provide a comprehensive overview of the replication cycle, pathogenesis, and long-term effects of SARS-CoV-2, particularly from the perspective of host factors and host-virus interactions. This knowledge would be key for the identification of new pathophysiological aspects and mechanisms, and the discovery of actionable therapeutic targets and strategies for COVID-19 and related diseases.
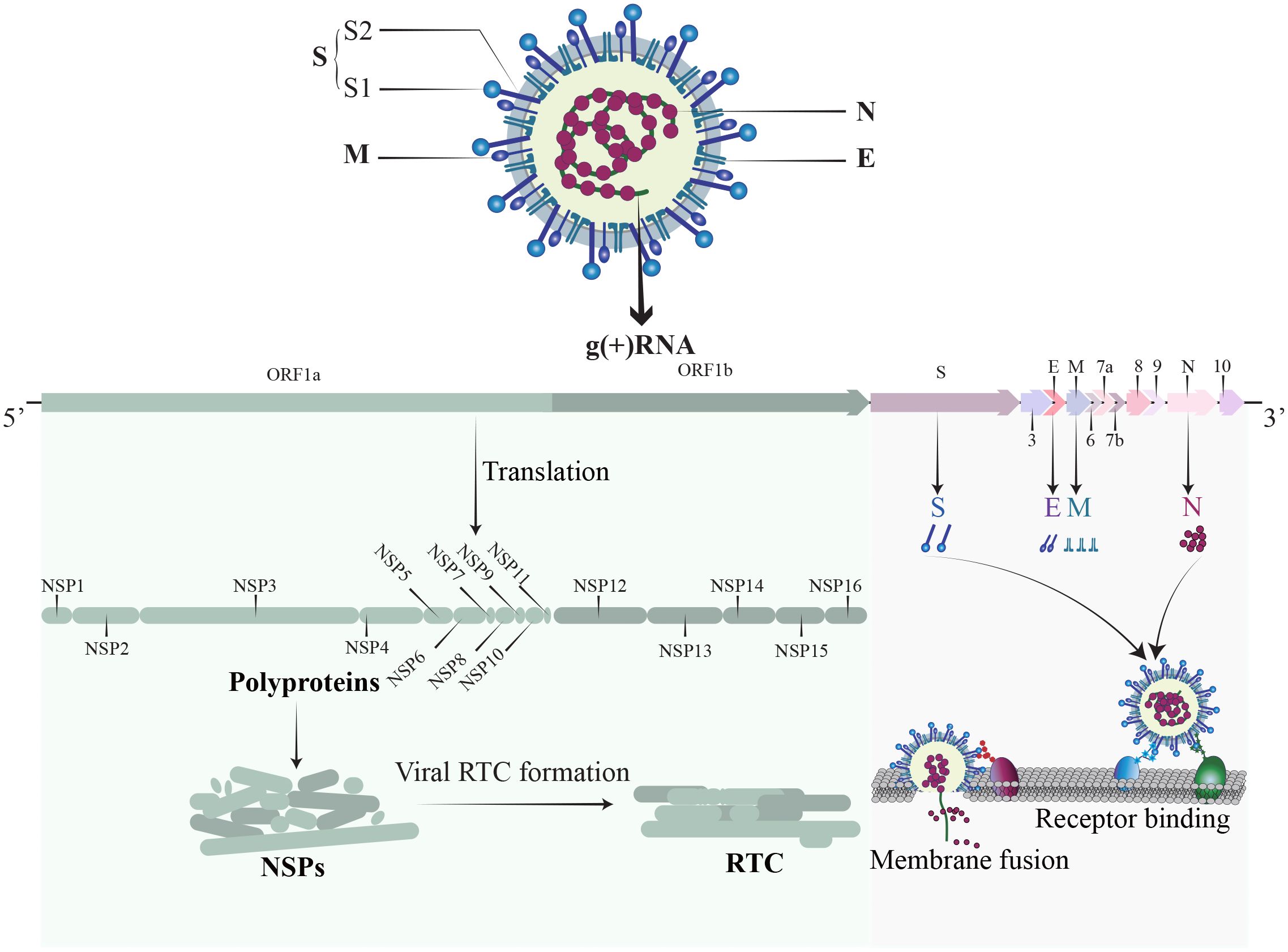
2 Structure of SARS-CoV-2SARS-CoV-2, a member of the Coronaviridae family, is an enveloped, positive-sense single-stranded RNA virus (Figure 1). The SARS-CoV-2 genome is approximately 30 kb and consists of at least 13 recognized open reading frames (ORFs) and 2 untranslated regions (UTRs) (Malone et al., 2022). The protein component of SARS-CoV-2 is composed of non-structural proteins (NSPs), structural proteins, and accessory proteins. NSPs are crucial for RNA-dependent RNA polymerase (RdRp) holoenzyme formation, replication organelle formation, and viral protein synthesis (Malone et al., 2022; Li et al., 2023a), and play a vital role in immune evasion and abnormal inflammatory response (Zhou et al., 2021). The structural proteins of SARS-CoV-2 include a nucleocapsid protein (N), a spike protein (S), a membrane protein (M), and an envelope protein (E) (Li and Chang, 2023). The N protein encapsidate the viral RNA genome and forms a protective nucleocapsid, shielding the viral RNA from cytoplasmic immune surveillance while facilitating the assembly of nucleoprotein complexes (Lu et al., 2011; Scherer et al., 2022). The S protein is vital for the entry of SARS-CoV-2 (Yu et al., 2023). Following enzymatic cleavage at the furin site, the S protein undergoes division into two subunits: S1 and S2 (Jackson et al., 2022). The S1 subunit forms the spike head and contains the receptor binding domain (RBD) that is primarily responsible for receptors recognition, and the S2 subunit attaches to the viral envelope and forms a stem-like structure that activates membrane fusion (Walls et al., 2020). The M protein is indispensable for virus assembly and contributes to the formation of viral envelope (Zhang et al., 2022b), while the E protein has a role in vesicle transport, budding, and intercellular transmission. The accessory proteins are recognized as crucial virulence factors in various pathogenesis pathways during SARS-CoV-2 infection, primarily contributing to immune evasion (Zandi et al., 2022). In short, the structure of SARS-CoV-2 is relatively simple, yet its nucleic acids and proteins play multiple and vital roles in viral replication and pathogenesis.
Figure 1 The structure of SARS-CoV-2. SARS-CoV-2 is an enveloped, positive sense single-stranded RNA virus. The RNA genome contains 14 ORFs and 2 untranslated regions. The proteins of SARS-CoV-2 comprise non-structural proteins (NSP1-16), structural proteins (S, M, N, E), and a set of accessory proteins, each serving its own specific function.
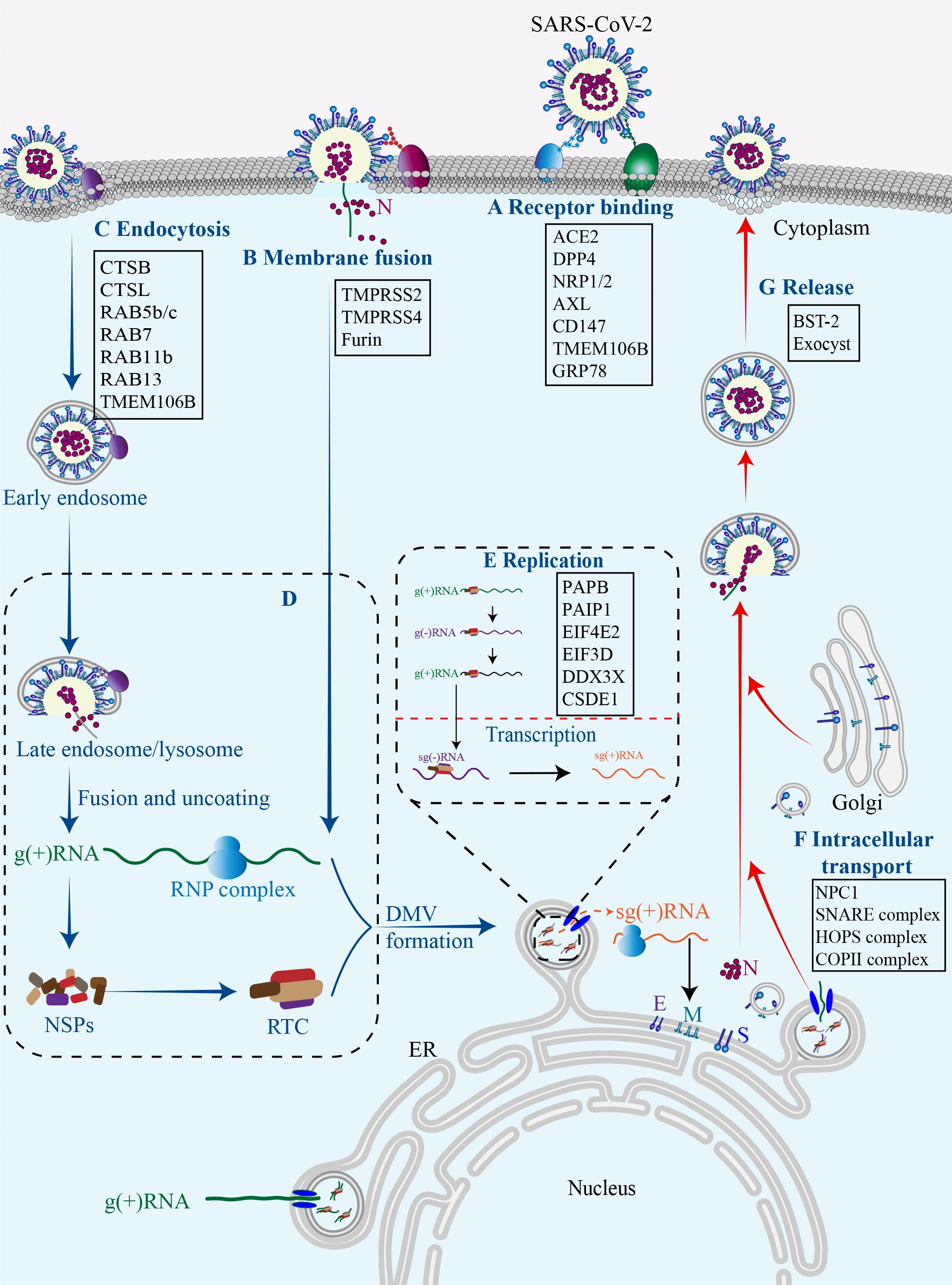
3 Host factors in SARS-CoV-2 replication cycleThe replication cycle of SARS-CoV-2 generally consists of the following stages: specific engagement of the S protein with host cell receptors; entry of virus into the host cell through membrane fusion or endocytosis; synthesis of viral RNA and proteins; assembly of progeny virions and their ultimate release (Chen et al., 2023b). Notably, the simple structure of SARS-CoV-2 alone is insufficient for completing its full replication cycle, and a number of host factors dynamically participate in each juncture of this intricate procedure (Figure 2) (Rajarshi et al., 2021).
Figure 2 Host factors in SARS-CoV-2 replication cycle.Numerous host factors are required for the completion of SARS-CoV-2 replication cycle. In brief, SARS-CoV-2 attaches to susceptible cells and the S protein binds to cell surface receptors (A). SARS-CoV-2 enters the cell through membrane fusion (B) or endocytosis (C). The virus releases its genetic material into the cell, and the viral gRNA initially functions as an mRNA and subsequently undergoes translation to generate NSP1-16, which then form the replication/transcription complex (D). In double-membrane vesicles, the viral gRNA serves as a template for replication and production of both gRNAs and sgRNAs. The latter functions as mRNA to synthesize structural proteins and other accessory proteins (E). The transportation of viral components within the cell occurs through the host membrane system (F). The newly synthesized gRNAs, structural proteins, and accessory proteins assemble into new virions which are subsequently released from infected cells via budding (G).
3.1 EntryThus far, there are two pathways responsible for the entry of SARS-CoV-2 into host cells, namely the membrane fusion pathway and the endocytosis pathway, both of which are initiated with the binding of the viral proteins to cell surface receptors. In both pathways, the S protein serves as an indispensable “promoter”. In the membrane fusion pathway, the S protein is cleaved on cell surface by host proteases, such as type II transmembrane serine protease (TMPRSS2), to assists the fusion of viral envelope with the cytoplasmic membrane (Glowacka et al., 2011). In the absence of cell surface proteases, SARS-CoV-2 may gain entry into host cells through endocytosis pathway. Within the endosome, cathepsin cleavage activates the S protein to initiate fusion between the viral envelope and the endosomal membrane (Ou et al., 2021). Upon cleavage and activation, the S protein undergoes conformational changes that result in the conversion from a fusion-competent state to a membrane-embedded homotrimeric prehairpin, followed by further transformation into a trimer-of-hairpin structure. During these transitions, the S protein exposes the fusion peptide, leading to the formation of fusion pores in the membrane through tight adhesion and semi-fusion, ultimately complete membrane fusion (White et al., 2008).
Cell surface receptors are essential for SARS-CoV-2 infection. Among them, angiotensin-converting enzyme 2 (ACE2) serves as the predominant receptor for SARS-CoV-2 entry, primarily by interacting with the RBD on the S1 subunit through its peptidase domain (Samelson et al., 2022). The binding mode of SARS-CoV-2 and ACE2 closely resembles that of SARS-CoV (Oudit et al., 2023). However, in comparison to SARS-CoV, the binding of SARS-CoV-2 S protein and ACE2 demonstrates more extensive interactions and increased affinity (Evans and Liu, 2021). In addition to ACE2, other cell surface receptors can also interact with the S protein and facilitate virus infection. Dipeptidyl peptidase 4 (DPP4), also known as CD26, is a transmembrane protein and the primary receptor for MERS-CoV, and thus has been considered a potential receptor for SARS-CoV-2. Indeed, DPP4 can interact with S protein RBD through its four residues (Mani et al., 2023). Interestingly, the membrane-anchored DPP4 is a virus receptor, while the soluble form (sCD26) acts as a competitive inhibitor to prevent the virus-DPP4 interaction (Raha et al., 2020). AXL, a tyrosine protein kinase receptor enriched in the lung and trachea, is another receptor for SARS-CoV-2 by interacting with the N-terminal domain (NTD) of the S protein (Fang et al., 2023). The co-localization of the S protein with AXL and endocytosis/vesicle transport markers in infected cells suggests that AXL may boost virus internalization through a clathrin-dependent mechanism (Wang et al., 2021c). The cleavage of the S protein by furin protease generates a binding site for neuropilin-1 (NRP1) and NRP2, indicative of potential SARS-CoV-2 receptors (Cantuti-Castelvetri et al., 2020). In addition, CD147 has been reported to interact with the S protein and facilitate the entry of SARS-CoV-2 through endocytosis (Wang et al., 2020b). Altogether, accumulating data suggest that the ACE2 receptor plays a pivotal role in facilitating SARS-CoV-2 cell entry, while other receptors may synergistically collaborate with ACE2 to enhance viral infectivity. However, the presence of certain receptors in cells lacking ACE2 expression expands the range of SARS-CoV-2 tropism, thereby rendering virtually all tissue cells susceptible to SARS-CoV-2 infection. It is of profound significance for future study to unravel these receptors and their action mechanisms.
Thus far, three cellular proteases are primarily involved in the hydrolytic activation of the S protein. Among them, TMPRSS2 plays an essential role in facilitating the fusion of the SARS-CoV-2 envelope with the host cell plasma membrane by cleaving the S protein at the S2’ sites and exposing the fusion peptide (Shapira et al., 2022). Interestingly, TMPRSS2 has been observed to co-localize with ACE2 in various cell types, including lung type II pneumocytes, ileal absorptive enterocytes, and nasal goblet secretory cells. It not only provides a reasonable explanation for the vulnerability of these cells to SARS-CoV-2, but also highlights the significance of TMPRSS2 as a pivotal host protease in viral invasion (Ziegler et al., 2020). Consistent with this, the absence of co-expression of ACE2 and TMPRSS2 in individual pancreatic β cells diminishes the probability of direct SARS-CoV-2 infection in vivo (Coate et al., 2020). Another serine protease called TMPRSS4 can also enhance S protein cleavage and facilitate virus entry. Although TMPRSS4 is less effective than TMPRSS2, its abundance in human intestinal epithelial cells may predominantly contribute to virus infection in the gastrointestinal tract (Zang et al., 2020). Furin protease, renowned for its ability to cleave envelope glycoproteins of influenza and human immunodeficiency virus (HIV) (Follis et al., 2006). can pre-cleave the S protein at the S1/S2 boundary, resulting in S1 and S2 subunits. The pre-cleavage enhances subsequent cleavage by TMPRSS2 and contributes to the high infectivity and tropism of SARS-CoV-2 (Essalmani et al., 2022). Insufficient expression of TMPRSS2 or failure of a virus-ACE2 complex to encounter TMPRSS2 can result in the internalization of SARS-CoV-2 through the clathrin-mediated endosomal/lysosomal pathway. The cleavage of the S2’ site can be conducted by Cathepsin L (CTSL) and CTSB, leading to the initial fusion between the viral envelope and endosomal membrane (Hoffmann et al., 2020; Takeda, 2022).
Although several host proteases are involved in SARS-CoV-2 infection, the activation of the S protein is predominantly promoted by TMPRSS2, and the fusion process occurring at the cell surface represents the most efficient mechanism for viral entry (Jackson et al., 2022). It implies that targeting the process of membrane fusion could be more effective in preventing viral infection compared to endocytosis (Ou et al., 2021). In other words, inhibitors focusing on the endocytosis pathway demonstrate limited efficacy in restricting SARS-CoV-2 infection, while a combined approach involving a TMPRSS2 inhibitor alongside an endocytosis pathway inhibitor holds potential advantages.
3.2 ReplicationUpon entering the cytoplasm, the SARS-CoV-2 genome initiates its replication program. The viral genomic RNA (gRNA) initially serves as messenger RNAs (mRNAs) for translation. ORF1a and 1b encode the polyproteins PP1a and PP1ab respectively, which are subsequently cleaved by viral proteases into 16 distinct NSPs. NSP1 is responsible for halting the translation of host mRNAs, while the remaining NSPs mainly form replication-transcription complexes (RTCs) to facilitate viral RNA synthesis (Lou and Rao, 2022). The replication of the viral genome occurs within double-membrane vesicles (DMVs) that are perinuclear membrane structures derived from the endoplasmic reticulum (ER) and induced by SARS-CoV-2 infection (Snijder et al., 2020). Within DMVs, the gRNA acts as a template for synthesizing negative strand RNA intermediates, which in turn serve as templates for synthesizing new positive strands. The SARS-CoV-2 genome produces a total of 10 distinct (+) RNAs, comprising one full-length gRNA and nine sub-genomic RNAs (sgRNAs) (Wyler et al., 2021). Transmembrane pores facilitate the transportation of the newly formed viral RNAs from the DMV to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) for subsequent translation (Wolff et al., 2020). The translation of sgRNAs in the cytoplasm produce SARS-CoV-2 structural proteins (S, E, M, and N) as well as accessory proteins (3a, 3c, 6, 7a, 7b, 8, and 9b) (Lee et al., 2021).
The hijack of host machinery by SARS-CoV-2 is crucial for viral RNA replication and protein synthesis (Fung and Liu, 2019), yet it significantly impairs the normal gene expression of the host. NSP1 is a critical virulence factor and contributes to the host translational shutoff at multiple stages (Frolov et al., 2023), NSP1 can interact with the heterodimer NXF1-NXT1, a receptor for host mRNA export on the nuclear pore complex, and inhibit the cytoplasmic translocation of host mRNAs (Zhang et al., 2021a). NSP1 may also disrupt the translation of host mRNAs by inserting its C-terminal domain into the mRNA channels of the ribosomal 40s subunit (Schubert et al., 2020). Moreover, NSP1 is able to promote the degradation of host mRNAs by cleaving their 5’-UTR, thereby accelerating cellular mRNA decay mediated by the 5’-3’ exoribonuclease 1 (Lou and Rao, 2022; Tardivat et al., 2023). NSP3, also known as the SARS-unique domain (SUD), enhances the affinity between polyA-binding protein (PAPB, an essential translation factor) and PABP-interacting protein 1 (PAIP1), and thus promotes protein synthesis of SARS-CoV-2 rather than the host (Lei et al., 2021b). SARS-CoV-2 also causes DNA damage and disrupts the DNA damage response, resulting in genomic instability. Specifically, ORF6 and NSP13 cause the degradation of DNA damage response kinase CHK1 through proteasomal and autophagic mechanisms respectively, leading to DNA damage. The N protein may interfere with damage-induced long non-coding RNAs (lncRNAs) to impair 53BP1 focal recruitment and DNA repair (Gioia et al., 2023). Recently, a comprehensive SARS-CoV-2-human protein-protein interactome reveals a physical interaction between ORF3a and zinc finger protein 579 (ZNF579), an uncharacterized human protein likely functioning as a transcription factor, suggesting a potential impact on ZNF579-modulating transcriptome (Zhou et al., 2023).
To facilitate gene replication and expression, the viral RNA recruits specific host RNA-binding proteins (RBPs) to form ribonucleoprotein (RNP) complexes. Recently, a deep learning tool was employed to identify SARS-CoV-2-interacting RBPs and revealed numerous candidates, including heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), hnRNPK, hnRNPU, U2 small nuclear RNA auxiliary factor 2 (U2AF2), interleukin enhancer binding factor 3 (ILF3), TIA1 cytotoxic granule associated RNA binding protein (TIA1), insulin like growth factor 2 mRNA binding protein 1 (IGF2BP1), and staphylococcal nuclease and tudor domain containing 1 (SND1) (Sun et al., 2021b). Indeed, depletion of TIA1, SND1 and IGF2BP1 significantly reduced the production of SARS-CoV-2 RNA in infected cells (Sun et al., 2021b). It is worth mentioning that ILF3 is an important protein required for interleukin-2-expressing T cells, and can also form complexes with other proteins, dsRNAs, small non-coding RNAs (ncRNAs), and mRNAs, to widely regulate gene expression, thereby participating in the occurrence and development of a variety of tumors (Jiang et al., 2021; Zhang et al., 2023). However, its role and mechanism in COVID-19 remain unclear.
Besides, eukaryotic translation initiation factor 4E type 2 (EIF4E2), EIF3D, DEAD-box helicase 3 X-linked (DDX3X), and cold shock domain containing E1 (CSDE1) have been identified as pro-viral RBPs (Hoffmann et al., 2021; Lee et al., 2021). In contrast, zinc finger CCCH-type containing, antiviral 1 (ZC3HAV1), tripartite motif containing 25 (TRIM25), poly (ADP-ribose) polymerase family member 12 (PARP12), shiftless antiviral inhibitor of ribosomal frameshifting (SHFL), and RNA binding motif protein 24 (RBM24) are probably anti-viral RBPs (Lee et al., 2021; Yao et al., 2023). For instance, ZC3HAV1, activated by TRIM25 through ubiquitination, exerts the anti-viral effect by promoting RNA degradation and inhibiting translation (Zheng et al., 2017), and SHFL may obstruct ribosomal coding during ORF1 translation (Schmidt et al., 2021). These findings suggest that the status of virus replication is partially dependent on the equilibrium and dynamic responses of pro- and anti-virus RBPs.
3.3 Transport and releaseAfter synthesis, the viral genome and proteins are transported to ERGIC for assembly, leading to the formation of complete progeny virions that are then released from the cell surface through exocytosis (Ravi et al., 2022). It is increasing known that this transportation is intricately linked to the host membrane system. Intriguingly, approximately 40% of the host proteins involved in the interaction with SARS-CoV-2 are associated with endomembrane compartments or vesicle trafficking pathways (Gordon et al., 2020). It is reasonable to speculate that the regulation of intracellular transport of viral components by host factors is likely to exert a significant impact on the viral replication cycle.
In the endocytosis pathway, SARS-CoV-2 is transported to early endosomes in a RAS-associated protein 5 (RAB5)-dependent manner through the interaction between the S protein and RAB5b/c (Chen et al., 2021). The S protein can also interact with RAB11b and RAB13, which are associated with early endosomes and regulate the trafficking of receptors and other ligands to the membrane. In late endosomes, the S protein may interact with late endosomal proteins, including RAB7a, RAB7b, and RAB7l1, to potentially facilitate lysosomal maturation (Tiwari et al., 2021). While in the cytoplasm, the M protein and the ORF7b possess the capability to interact with several components of the soluble NSF attachment protein receptor (SNARE) complex, an essential complex for all membrane fusion process, such as syntaxin 6 (STX6) and STX10. Additionally, the ORF3a interacts with the homotypic fusion and protein sorting (HOPS) complex, which regulates membrane vesicle transport through mechanisms involving membrane fusion. Furthermore, NSP13 may interact with the Golgi complex (Gordon et al., 2020), while the ORF6 protein binds to components of the coatmer protein II (COPII) complex that is crucial for vesicle formation and substance transportation from the endoplasmic reticulum to the Golgi apparatus (Chen et al., 2021). In short, the interactions between virus and host proteins maximize their intracellular transportations. Among them, lysosomal transmembrane protein 106B (TMEM106B) is a prominent representative (Wang et al., 2021b). TMEM106B plays important roles in lysosomal transport, acidification, and lysosomal enzyme expression, and it is thus reasonable that it can regulate viral infection through these actions (Luningschror et al., 2020). Moreover, it has recently reported that TMEM106B may function as an alternative receptor for SARS-CoV-2 entry into ACE2-negative cells and facilitate the formation of spike-mediated syncytia (Baggen et al., 2023), further expanding its implication in SARS-CoV-2 pathogenesis. The exocyst, identified through CRISPR screens, is an octameric protein complex that facilitates the tethering of secretory vesicles to the plasma membrane. Consequently, this complex may enhance viral particle trafficking during entry or egress and regulate surface expression of viral entry factors (Mei and Guo, 2018; Wang et al., 2021b). It has been proved that SARS-CoV-2 infection efficiency was regulated by EXOC2, one of key subunits of the exocyst complex (Yi et al., 2022).
There are also host proteins that play a role in the release phase of the virus. Bone marrow stromal antigen 2 (BST-2), also known as tetherin, is found in the plasma membrane, trans-Golgi network, and circulating endosomes, and can inhibit virus release (Tiwari et al., 2019). Mechanistically, BST-2 interacts with the S protein and thus forms a bridge between the budding virions and the plasma membrane, tethering the new virions to the plasma membrane to inhibit their release (Tiwari et al., 2019, Tiwari et al, 2021). However, this anti-viral activity can be impeded by ORF7a that directly binds to BST-2 and hinders its glycosylation process (Mann et al., 2023).
During SARS-CoV-2 infection, viral proteins extensively remodel host cell endomembrane through interactions with host factors to facilitate various stages of the viral cycle, Including the formation of DMVs for viral replication and the modulation of the lysosome pathways for virus release. The endomembrane system is of great potential to serve as a therapeutic target against SARS-CoV-2, its emerging variants, and even novel coronaviruses.
4 Host genetic factors in SARS-CoV-2 susceptibility and COVID-19 severityIt is well established that the severity and fatality risk of COVID-19 are intricately linked to the viral load of SARS-CoV-2 (Fajnzylber et al., 2020; El Zein et al., 2021; Tarafder and Khan, 2023), which in turn may be influenced by host gene polymorphism (Roy-Vallejo et al., 2023). Consequently, recent research highlights that the heterogeneity observed in the clinical manifestation of acute SARS-CoV-2 infection may be attributed, at least partially, to human genetic variation (Redin et al., 2022). This notion is highlighted by ACE2, the pivotal receptor for SARS-CoV-2. Two ACE2 variants are associated with increased SARS-CoV-2 susceptibility: the minor A allele in the rs2106806 variant and the minor T allele in the rs6629110 variant, with the later leading to a substantial increase in ACE2 expression (Martinez-Sanz et al., 2021). An in vivo study in hamsters suggests that higher ACE2 expression was associated with a higher viral load and more intense post-acute sequelae (Delval et al., 2023). Genetic polymorphism of ACE2 affect not only the expression of ACE2, but also the affinity between ACE2 and the S protein (Chan et al., 2020). The interaction between the S protein and ACE2 can be increased by certain single nucleotide polymorphisms (SNPs), such as rs73635825 and rs1244687367, but decreased by others, including rs1348114695 and rs1192192618 (Calcagnile et al., 2021; Lanjanian et al., 2021; Suryamohan et al., 2021).
Polymorphisms in other host genes, such as TMPRSS2, DPP4, interferon-lambda-3, tolloid like-1, discoidin domain receptor 1 and HLA-DRB1*08, may also contribute to the susceptibility and severity of COVID-19 (Irham et al., 2020; Senapati et al., 2020; Agwa et al., 2021; Amoroso et al., 2021). A meta-analysis revealed a significant correlation between TMPRSS2 rs12329760 C-allele and an elevated risk of developing severe COVID-19 (Saengsiwaritt et al., 2022). This result may be explained by positive correlation between TMPRSS2 activity and viral load (Okugawa et al., 2023). A genome-wide association study (GWAS) identified that a variant on chromosome 5 at 5q35 (rs60200309-A), close to the dedicator of cytokinesis 2 gene (DOCK2), is associated with severe COVID-19 in patients less than 65 years of age (Namkoong et al., 2022). In addition, genetic variants, such as KLRC2 deletion and reduced HLA-E*0101 levels, have shown to be risk factors for severe COVID-19 (Vietzen et al., 2021). Given the emerging role of genetic factors in the discrepancies in individual susceptibility and severity, genetic predisposition has been considered to a key modulator for SARS-CoV-2 pathogenesis.
In addition to genetic predisposition, the expression of host factors is dynamically and strictly regulated by numerous factors, which might in turn affect disease progression, adding an additional complexity to SARS-CoV-2 pathogenesis. For example, BRD2, an important regulator for ACE2 expression in human lung epithelial cells and cardiomyocytes, has demonstrated to be crucial for the host response to SARS-CoV-2. Correspondingly, BRD2 inhibitors effectively reduced endogenous ACE2 expression and prevented SARS-CoV-2 infection in these cells (Samelson et al., 2022). High mobility group box 1 protein (HMGB1), an evolutionarily conserved chromatin-binding protein that regulates ACE2 expression, is essential for the viral entry of both SARS-CoV-2 and SARS-CoV (Wei et al., 2021). Interestingly, a negative correlation between ACE2 expression and COVID-19 severity has been proposed (Mariappan and Balakrishna, 2020; Ortatatli et al., 2023). The large GTEx data revealed that ACE2 expression is high in Asian females and young people, those who are known to be less susceptible, and even less inflicted by severe or fatal outcome, while it is low in males, further decrease with age and type 2 diabetes mellitus (T2DM), those who are most susceptible to bad outcome (Chen et al., 2020a). The decreased ACE2 expression in males, older individuals, and T2DM patients might contribute to their poor prognosis and high lethality. This phenomenon is likely attributed to the conversion of angiotensin II (Ang-II) to Ang 1-7 by ACE2, which reduces the detrimental effects of Ang-II (Donoghue et al., 2000).
Although GWAS has identified a variety of common genetic variants associated with COVID-19, the functions and mechanisms of these variants are still in the nascent phase. By revealing the genes and pathways involved in SARS-CoV-2 pathogenesis and host-virus interactions, human genome research would not only uncover novel targets for prevention and therapy, but also lay the groundwork for more personalized disease management.
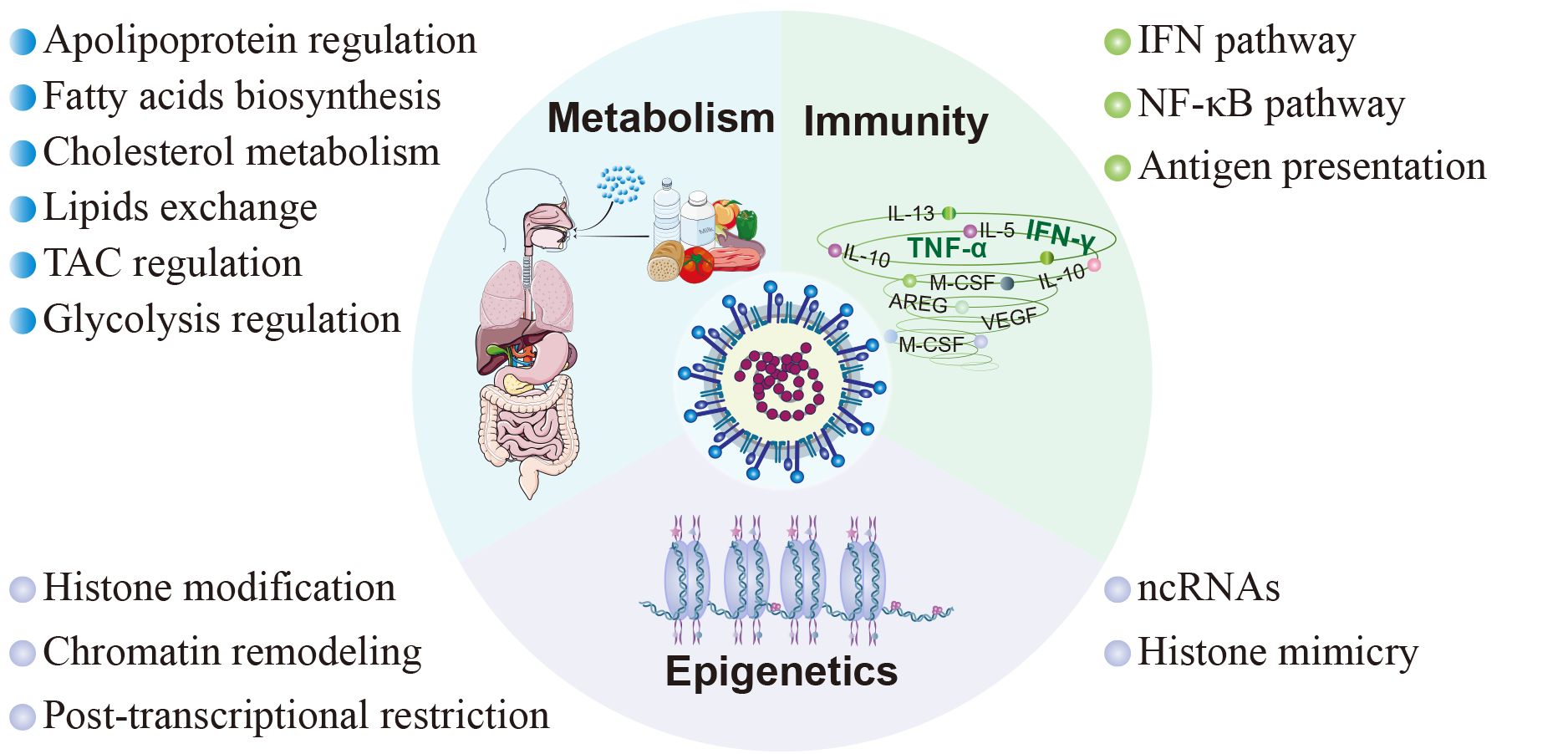
5 Host factors in SARS-CoV-2-triggered responsesThe clinical manifestations of SARS-CoV-2 infection vary from asymptomatic or mild/moderate upper respiratory tract symptoms to life-threatening COVID-19 pneumonia with multi-organ complications (Melenotte et al., 2020). These diverse clinical consequences are closely associated with the host response to SARS-CoV-2, encompassing immune responses, metabolic alterations, and epigenetic modifications (Figure 3).
Figure 3 Host factors in SARS-CoV-2 pathogenesis. The pathogenesis subsequent to viral infection involves diverse facets of the host inherent physiological processes, encompassing immunity, metabolism, and epigenetics. In terms of immunity, SARS-CoV-2 modulates the innate immune response through the interferon signaling pathway and NF-κB signaling pathway, and inhibits adaptive immunity by interfering with antigen presentation. As to metabolism, SARS-CoV-2 interferes with cellular metabolism by participating in the regulation of cholesterol metabolism, fatty acid production, tricarboxylic acid cycle, glycolysis, and cell membrane lipid exchange. Moreover, SARS-CoV-2 disrupts host cell epigenetics by means of non-coding RNAs, histone mimicry, chromatin remodeling, and some other mechanisms.
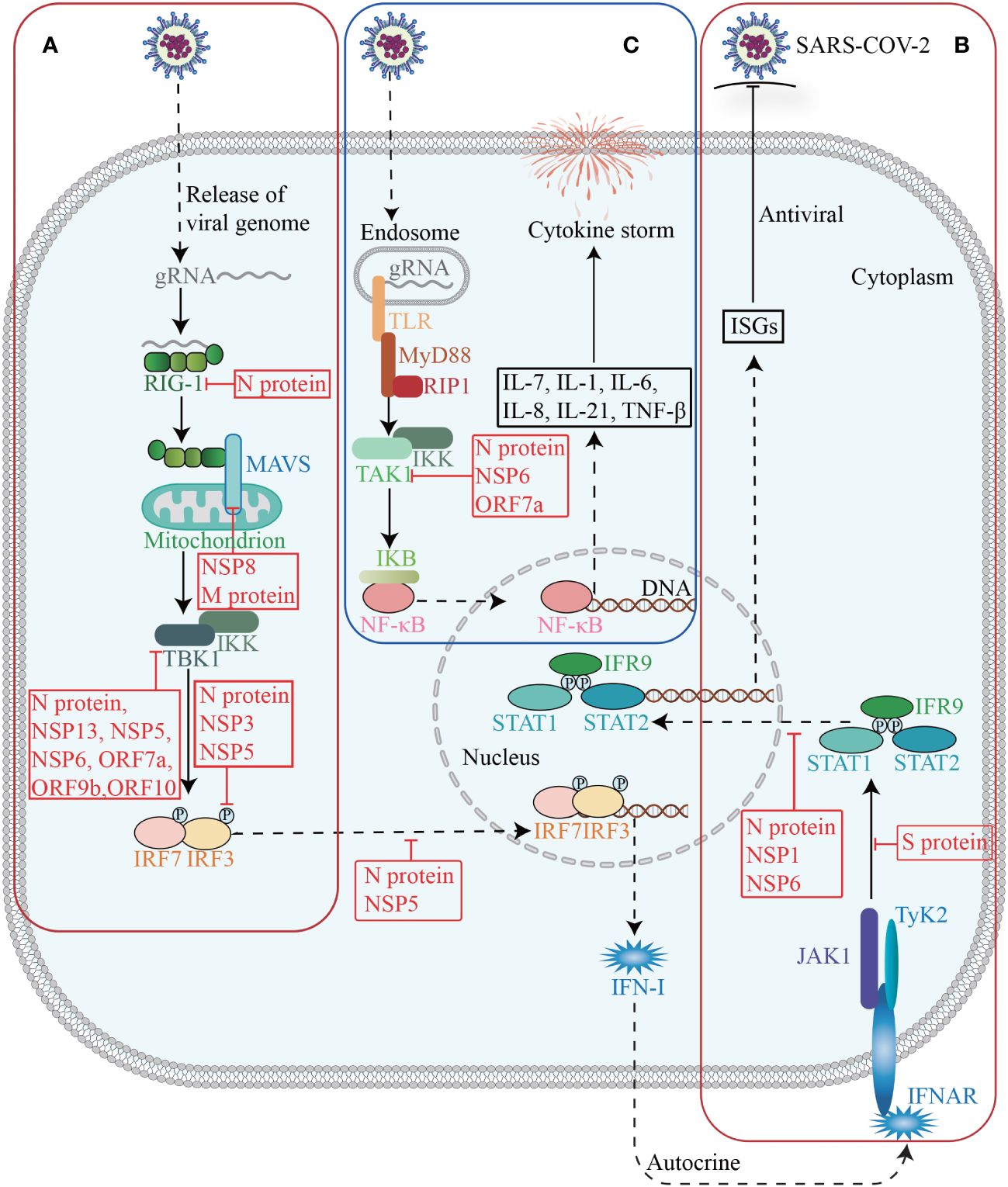
5.1 Immune responsesThe innate immune system serves as the primary defense against SARS-CoV-2, and plays a crucial role in monitoring viral infections, eliminating virus-infected cells, initiating inflammation, and enhancing adaptive immunity. Conversely, SARS-CoV-2 could promote immune evasion through interactions with host proteins, thereby limiting host control and helping itself to replicate and transmit (Diamond and Kanneganti, 2022) (Figure 4).
Figure 4 Host factors in immune response. The invasion of SARS-CoV-2 triggers the activation of innate immunity, which subsequently induces the synthesis and secretion of IFN-I (A). Upon binding to its receptor, IFN-I activates the JAK-STAT signaling pathway and subsequently induces the expression of ISGs (B). Activation of TAK1 and IKK triggers the initiation of the NF-κB pathway, leading to a cascade of downstream cytokines and chemokines (C).
The interferon (IFN) signaling pathway is an essential innate immune defense mechanism against SARS-CoV-2. Upon SARS-CoV-2 infection, signaling cascades are triggered through the recognition of pathogen-associated molecular patterns (PAMPs) by pattern-recognition receptors (PRRs), leading to the production of type I IFN and type III IFN (IFN-I and IFN-III) and the activation of IFN signaling pathways in an autocrine and paracrine manner (Akira et al., 2006; Minkoff and tenOever, 2023), ultimately inducing hundreds of interferon-stimulated genes (ISGs) with various antiviral functions to foster an antiviral state (Schultze and Aschenbrenner, 2021).
Many studies have shown that SARS-CoV-2 employs a multifaceted strategy to suppress the production of IFN-I and IFN-III (Minkoff and tenOever, 2023). Patients with severe and critical COVID-19 have a highly impaired IFN-I response (characterized by the absence of IFN-β and low IFN-α production and activity), which is associated with persistent blood viral load and increased inflammation (Hadjadj et al., 2020). From a holistic view, SARS-CoV-2 could suppress immune responses, by modulating host gene expression, with its NSPs and accessory proteins as essential executors. As mentioned above, NSP1 can widely repress host gene expression through multiple ways. Recent evidence suggests that NSP16 is capable of suppressing host mRNA splicing, whereas NSP8 and NSP9 could interfere protein trafficking to the cell membrane (Banerjee et al., 2020). By interacting with the nuclear pore complex component NUP98-RAE1, ORF6 can either inhibit mRNA nuclear export and thus reshape the host cell proteome, or interfere with nuclear import, specifically the translocation of IRFs and STATs, thereby disrupting IFNs antagonism (Kehrer et al., 2023). These mechanisms independently act and coordinately decrease the expression of host genes, especially IFN-I and IFN-III.
In addition, SARS-CoV-2 NSPs and accessory proteins may interact with essential proteins involved in the production and signaling of IFNs, thereby counteracting their antiviral effects. NSP1 significantly inhibits the phosphorylation and translocation of signal transducer and activator of transcription 1 (STAT1), and thus suppresses the IFN-I signaling (Xia et al., 2020). NSP3 exhibits multifunctionality by cleaving the polypeptide chain translated from the SARS-CoV-2 genome, as well as mediating the removal of polyubiquitin chains and IFN-stimulated gene 15 (ISG15, a ubiquitin-like modifier) from host proteins (Klemm et al., 2020). The latter allows the virus to evade the defenses of the innate immune system. For example, NSP3 can antagonize the conjugation of ISG15 to impair the activation of melanoma differentiation gene 5 (MDA5) (Liu et al., 2021). The deubiquitylation of STING by NSP3 may disrupt the STING-inhibitor of NF-κB kinase-ϵ (IKKϵ)-interferon regulatory factor 3 (IRF3) complex, which is responsible for inducing the production of IFN-β as well as IFN-stimulated cytokines and chemokines (Cao et al., 2023). NSP5 is able to inhibit IFN-I and IFN-III expression by inhibiting the phosphorylation of TRAF family member-associated NF-κB activator binding kinase 1 (TBK1) and IRF3, as well as restraining IRF3 translocation (Zheng et al., 2022). NSP6, NSP13, and ORF9b may directly bind to TBK1 and thus impair phosphorylation-mediated TBK1 activation, while ORF7a can decrease TBK1 expression (Minkoff and tenOever, 2023). Besides, the interaction between ORF10 and STING attenuates the association between STING and TBK1, thereby inhibiting cGAS-STING-induced IRF3 phosphorylation and translocation (Han et al., 2022). Furthermore, NSP8 has shown to disrupt the MDA5-mitochondrial antiviral signaling protein (MAVS) signalosome, resulting in a significant reduction of K63-linked polyubiquitination of MDA5 (Rashid et al., 2022).
SARS-CoV-2 structural proteins are also involved in suppressing the host IFN response. The N protein can competitively bind to TRIM25, an E3 ubiquitin ligase that facilitates RIG-I ubiquitination, and thus attenuate retinoic acid inducible gene I (RIG-I) activation and IFN-I and IFN-III production (Gori Savellini et al., 2021; Minkoff and tenOever, 2023). Besides, the N protein may target RIG-I cofactors, such as G3BP1 and PACT, and quarantine their binding to RIG-I. Furthermore, the N protein is able to inhibit IFN-I and IFN-III expression by suppressing TBK1 and IRF3 phosphorylation and restraining the nuclear translocation of IRF3 (Zheng et al., 2022). Similarly, the M protein has been reported to disrupt the ability of MAVS to establish the necessary scaffolding required for downstream transcription factor activation and to inhibit the function of additional host factors involved in MAVS signaling such as TBK1 (Minkoff and tenOever, 2023). In suppressing IFN signal, the fragments generated through caspase-6 cleavage of the N protein possess the ability to inhibit the production of IFN-β and decrease the expression of representative ISGs, such as interferon induced protein with tetratricopeptide repeats 3 (IFIT3) and 2’-5’-oligoadenylate synthetase 1 (OAS1) (Chu et al., 2022). Both N proteins and NSP6 suppress the phosphorylation of STAT1 and STAT2 as well as the nuclear translocation of STAT1 (Mu et al., 2020; Bills et al., 2023). The S protein can interact with STAT1 to impede its association with JAK1 (Zhang et al., 2021b).
The protective role of IFNs responses in acute SARS-CoV-2 infection is evident, whereas a limited and delayed IFNs response results in heightened production of proinflammatory cytokines and lung parenchymal injury due to infiltration of inflammatory cells. However, there is interpatient variability in the IFN response among individuals with COVID-19, with severe patients exhibiting prolonged production of IFN-I in certain cases and absence of IFN-I expression in others (Lowery et al., 2021). Therefore, while IFN therapy may not confer benefits to all patients, it holds potential utility for specific patient subsets, including those who receive early treatment during infection and individuals exhibiting markedly low IFN responses (Feld et al., 2021; Monk et al., 2021). Further investigations are warranted to reconcile these contradictory findings and establish a universally applicable approach for suitable populations amenable to IFN therapy.
The pathogenesis of COVID-19, particularly in severe cases, is characterized by an excessive systemic inflammatory response known as a cytokine storm, which is closely associated with the activation of the NF-κB signaling pathway (Hadjadj et al., 2020; Kim et al., 2023). Upon activation of PRRs, the adaptor protein myeloid differentiation primary response gene 88 (MyD88) is engaged to initiate the NF-κB pathway, ultimately resulting in the expression of a multitude of cytokines and chemokines (Akira et al., 2006).
SARS-CoV-2 can interact with crucial proteins within the NF-κB pathway, and exacerbate systemic inflammatory response. During viral assemble, after binding to viral RNA, the N protein undergoes robust liquid-liquid phase separation (LLPS), which recruits and enhances the association between the TAK1 and IKK complex, thereby promoting virus-triggered activation of NF-κB signaling (Wu et al., 2021b). NSP6 and ORF7a interact with TAK1 to facilitate NF-κB activation through K63-linked polyubiquitination by TRIM13 and RNF121 (Nishitsuji et al., 2022). NSP14 interacts with inosine-5’-monophosphate dehydrogenase 2 (IMPDH2) and promotes the nuclear translocation of NF-κB p65 (Li et al., 2022a). It is noteworthy that ORF8 can interact with IL-17 receptor A (IL17RA) to regulate the systemic IL-17 signaling pathway, which plays a pivotal role in recruiting monocytes and neutrophils and triggering a cascade of downstream cytokines and chemokines, including IL-1, IL-6, IL-8, IL-21, and tumor necrosis factor beta (TNF-β) (Liu et al., 2017). However, a recent study has provided additional evidence that ORF8 transmits inflammatory signaling in monocytes and macrophages through MyD88 independently of the IL-17R (Ponde et al., 2023).
In addition to the innate immune response, the antiviral defense also involves adaptive immunity, which encompasses B cells and T cells. CD8+ T cells acting as cytotoxic effector cells to eliminate virus-infected host cells. CD4+ T cells can be activated and differentiated into various subsets, exerting commanding and regulatory roles in modulating the activity of other immune cells such as B cells, CD8+ T cells and macrophages, while orchestrating a comprehensive range of immune responses through their production of cytokines and chemokines (Zhu and Paul, 2008). Adaptive immunity can also be compromised by SARS-CoV-2. Recent evidence has revealed a notable absence of lymph node and splenic germinal centers, as well as Bcl-6+ B cells in COVID-19. Additionally, there are also impaired Bcl-6+ T follicular helper cell generation and differentiation, imbalanced T-helper cell polarization, and heterogeneous CD8+ T cells maturation and polarization (Kaneko et al., 2020; Gil-Etayo et al., 2021; Kudryavtsev et al., 2022). Mechanistically, a recent study suggests that viral protein ORF8 could impair the activation of T cells. ORF8 could sequester MHC-I during its transit through the ER, which may hinder viral antigen presentation on the cell surface and reduce recognition and elimination of virus-infected cells by facilitating MHC-I degradation via autophagosome and autolysosome pathways (Zhang et al., 2021d).
These findings clearly demonstrate that SARS-CoV-2 not only hampers the IFNs signaling pathway and antigen presentation to achieve immune evasion, but also triggers the NF-κB pathway, resulting in a severe inflammatory state. Some studies have suggested that the progression and severity of the disease during SARS-CoV-2 infection may be account for these two opposite immune abnormalities. That is, in the early stage of COVID-19, a variety of viral proteins inhibit immune signaling pathways, while in the late stage, the immune response is activated to a certain extent by specific viral proteins, leading to the occurrence of cytokine storm syndrome in patients with severe COVID-19 (Tian et al., 2020). The diverse effects of viral proteins during different periods may be attributed to variations in their proportions (Blanco-Melo et al., 2020), necessitating further investigation into the specific underlying mechanisms.
5.2 EpigeneticsRecent findings have suggested that SARS-CoV-2 significantly disrupts epigenetic regulation of host cells through ncRNA, histone mimicry and modification, chromatin remodeling and post-transcriptional restriction (Figure 3) (Kee et al., 2022). Among them, ncRNAs, particularly lncRNAs and microRNAs (miRNAs), are the most extensively studied, and can exert regulatory control over immune mechanisms, cellular damage, and virus-related physiological processes by modulating host proteins or viral genomes (Zhang et al., 2021c) (Table 1).
Table 1 miRNAs in SARS-CoV-2 pathogenesis. .
The infection of SARS-CoV-2 triggers an upregulation of host miRNAs, which subsequently targeting numerous host genes. The S protein stimulates the generation and release of exosomes containing miR-148a and miR-590, which are internalized by microglia to inhibit the expression of ubiquitin specific peptidase 33 (USP33) and downstream IRF9 in the nervous system. These miRNAs play a role in the regulation of TNFα, NF-κB, and IFN-β pathways, resulting in excessive activation of microglia and subsequent damage to the central nervous system (Mishra and Banerjea, 2021). In cardiomyocytes, miR-200c plays an antiviral role in regulating the entry of SARS-CoV-2 by targeting the 3’-UTR of ACE2 mRNA and subsequently reducing its expression (Lu et al., 2020). In endothelial cells, miR-98-5p directly targets and represses TMPRSS2 (Matarese et al., 2020). RNA-seq and bioinformatics analysis reveal extensive complementarity between multiple human miRNAs and the functional RNAs of SARS-CoV-2 (Sacar Demirci and Adan, 2020; Hill and Lukiw, 2022), such as miR-447b and the RNA of the S protein, miR-3672 and the RNA of the E protein, and miR-325 and the RNA of the M protein. However, the epigenetic regulation of viral proteins by human miRNAs still needs further investigation and verification. The expression profiles of human miRNAs exhibit significant tissue-specific and inter-individual variations (Hill and Lukiw, 2022). The tissue-specific expression patterns of miRNAs may partially contribute to the variations in susceptibility and pathological response to SARS-CoV-2 across different tissues, while the significant differences in miRNA abundance and functionality among individuals may result in disparities in immune response and disease severity.
On the other hand, SARS-CoV-2 also encode its own miRNAs. miRNAs derived from SARS-CoV-2 have the potential to modulate the host immune system and inflammatory response (Zhang et al., 2021c). For instance, MR385-3p may could bind to 5’-UTR of TGFBR3, a key receptor of immune system, and MR66-3p may could bind to the enhancer of TNF-α, an important cytokine in the cytokine storm. While a viral miRNA-like small RNA (CoV2-miR-O7a) has been identified to target basic leucine zipper ATF-like transcription factor 2 (BATF2), thereby modulating the IFN signaling (Pawlica et al., 2021). In addition, MD25p and MR147-3p target the apoptosis-related gene CHAC1 and RAD9A respectively, involved in the host cellular apoptotic response to viral infection (Zhang et al., 2021c). Given the important role of host and viral encoded miRNAs in COVID-19 pathogenesis, the exploration for specific miRNAs with potential therapeutic benefits against SARS-CoV-2 can be regarded as a promising strategy to mitigate the severity of COVID-19. However, since a single miRNA can regulate a large number of mRNAs, high doses of a single miRNA in vivo might lead to severe off-target adverse effects. Therefore, it may be an effective therapeutic strategy to clarify the target of various miRNA and use multiple low-dose miRNAs in combination.
LncRNAs can modulate gene regulation at multiple levels, including chromatin remodeling, transcriptional regulation, and post-transcriptional processing Ernst and Morton, 2013). An increasing number of lncRNAs have been implicated in SARS-CoV-2 infection and subsequent host responses (Qiu et al., 2018). For example, the expression of IL-6 can be regulated by several lncRNAs, including TSLNC8, MALAT1, NEAT, CAIF and HOTAIR, through various pathways such as JAK/STAT, NF-κB, hypoxia inducible factor-1α (HIF-1α), and mitogen-activated protein kinase (MAPK) (Serpeloni et al., 2021). More recently, it has shown that the SARS-CoV-2 N protein hinders the recruitment of DNA damage response factor 53BP1 at DNA double-strand breaks by competing with the binding of damage-induced lncRNAs, and thus impedes DNA repair, adding another complexity to lncRNA regulation on SARS-CoV-2 pathogenesis (Gioia et al., 2023). Notably, a multitude of lncRNAs are differentially expressed in response to viral infection. A deeper understanding of the change in lncRNA transcriptome in infected cells, as well as their effects on interactions between host and virus, could eventually facilitate the identification of novel therapeutic targets and the development of new diagnostic biomarkers and therapeutic treatments.
In addition to ncRNAs, SARS-CoV-2 can impact other forms of epigenetic modifications. ORF8 functions as a histone mimic of the ARKS motifs in histone H3 to disrupt host cell epigenetic regulation (Kee et al., 2022). NSP5 can interact with histone deacetylase 2 (HDAC2) to modulate HDAC2 nuclear entry and subsequent inflammation and IFN response (Xu et al., 2019; Gordon et al., 2020). The E protein may bind to bromodomain containing 2 (BRD2) and BRD4 (Gordon et al., 2020). and thus influence their interaction with acetylated histones and gene transcription (Faivre et al., 2020). Interestingly, a multitude of transcription factors, such as JUN, and zinc finger and BTB domain containing 20 (ZBTB20), are responsible to SARS-CoV-2 infection (Alexander et al., 2021). It is evident that post-transcriptional restriction of JUN can impede the induction of IFNs, while ZBTB20 reduction may delay the immune response. These findings suggest that SARS-CoV-2 hampers the host’s immune response through diverse epigenetic regulatory mechanisms.
5.3 MetabolismThe prediction of protein targets for SARS-CoV-2 reveals a significant enrichment in metabolic processes related to lipids, amino acids, and glucose (Dey et al., 2020), suggesting a profound impact on host metabolism.
In addition to be the main receptor for SARS-CoV-2, ACE2 can regulate the expression of many critical genes involved in glucose and lipid metabolism, such as glucose-6-phosphatase, catalytic (G6PC), glucose transporter 2 (GLUT2), peroxisome-proliferator-activated receptor coactivator (PGC)-1α, peroxisome proliferators-activated receptors α (PPARα), and PPARγ, suggesting a role in SARS-CoV-2-induced metabolic dysregulation (Li et al., 2022b). Recently, SARS-CoV-2 infection has been shown to increase the expression of RE1-silencing transcription factor (REST), which regulates hundreds of genes including many metabolic factors, eventually leading to abnormal metabolism of glucose and lipids (He et al., 2021).
Genome-wide CRISPR knockout screening studies suggest cholesterol metabolism as the most significant pathways associated with SARS-CoV-2 infection (Schneider et al., 2021). Several lines of evidence indicate that cholesterol may facilitate viral infection. Putative cholesterol recognition amino acid consensus motifs have been recently identified in the S protein, and antibodies that block these cholesterol-binding sites significantly hindered SARS-CoV-2 entry (Wei et al., 2020; Baier and Barrantes, 2023). Amlodipine, the drug that disrupts the cholesterol biosynthesis pathway, is sufficient to reduce SARS-CoV-2 infection (Daniloski et al., 2021). Similarly, 25-hydroxycholesterol that activates acyl-coA: cholesterol acyltransferase (ACAT) and leads to cholesterol depletion on the plasma membrane can prevent SARS-CoV-2 entry (Wang et al., 2020c). Niemann-Picker intracellular cholesterol transporter 1 (NPC1), a regulator of endosomal/lysosomal vesicle transport and cholesterol efflux, is important for the cytoplasmic entry of the viral genome, and its mutations or deletions exhibit an anti-viral effect (Vial et al., 2021). The high-density lipoprotein (HDL) components could bind to S1 subunit of the S protein, and thus enhance the HDL scavenger receptor B type 1 (SR-B1)-mediated virus attachment on cell surface and subsequent virus uptake (Wei et al., 2020). Transmembrane protein 41B (TMEM41B) may contribute to the formation of viral replication complex via mobilization of cholesterol and other lipids to facilitate host membrane expansion and curvature (Ji et al., 2022). Interestingly, apolipoprotein E (APOE), a key factor in cholesterol metabolism, could interact with a key docking site for SARS-CoV-2 S protein on ACE2, thereby reducing ACE2/S-mediated viral entry into cells. Taken together, as a conserved cellular mechanism exploited by viruses for host cell entry and replication, the lipid metabolism pathway represents a crucial research avenue in the development of host-targeted antiviral drugs.
From another aspect, SARS-CoV-2 infection disrupts normal lipid metabolism. General dyslipidemia was observed by targeted or untargeted lipidomic analyses of plasma, particularly in severe COVID-19 patients (Casari et al., 2021). These abnormally metabolized lipids not only have a structural role, but also regulate numerous signaling pathways and immune responses (Hannun and Obeid, 2018). Mechanistically, SARS-CoV-2 could interact with sterol regulatory element binding transcription factor chaperone (SCAP), and membrane-bound transcription factor peptidase, site 1 and 2 (MBTPS1 and MBTPS2), as well as regulators of the cholesterol biosynthesis pathway, to modulate the biosynthesis of fatty acids and cholesterol (Wang et al., 2021b). The viral protein NSP6 can upregulate many cholesterols metabolism-related proteins, such as 7-dehydrocholesterol reductase, cytochrome p450 family 51 subfamily A member 1, isopentenyl-diphosphate delta 1somerase 1, and squalene epoxidase (Stukalov et al., 2021). Besides, the S protein has the ability to extract lipid components from HDL, leading to a modification in the HDL functionality for lipid exchange with model cellular membranes (Correa et al., 2021). As strongly altered in COVID-19 and correlate with the severity of the disease, specific lipid components may serve as potential biomarkers for the identification and severity classification of COVID-19.
Glucose metabolism is also disrupted by SARS-CoV-2 infection. COVID-19 can result in new-onset hyperglycemia or worsening glycemic control (Conte et al., 2024), and fasting plasma glucose levels can even predict the mortality of COVID-19 patients (Cai et al., 2020). Consistently, SARS-CoV-2 infection is also influenced by the host glucose metabolism. Blood glucose levels appear to be positively associated with viral load (Codo et al., 2020; Zhu et al., 2020; Garreta et al., 2022). Elevated blood glucose levels can increase the stability of ACE2 mRNA, leading to up-regulation of ACE2 expression and promoting the entry of SARS-CoV-2 into cells (Garreta et al., 2022; Vargas-Rodriguez et al., 2022). Like lipid metabolism, host glucose metabolism is regulated by SARS-CoV-2. For example, the interaction between NSP14 and sirtuin 5 facilitates the succinylation of host proteins including several key enzymes of the tricarboxylic acid cycle (TAC), thereby inhibiting cellular metabolic pathways (Liu et al., 2022c). SARS-CoV-2 also modifies proteins related to glycolysis. Although the underlying mechanisms remain unclear, SARS-CoV-2 infection leads to an excessive activation of glycolysis, resulting in a significant consumption of glucose, pyruvate, glutamine, and alpha-ketoglutaric acid. Notably, the blockade of glutaminolysis has been shown to impair both viral replication and the inflammatory response (de Oliveira et al., 2022).
Altogether, glucose and lipid metabolism are essential for the cell to conduct fundamental physiological functions. The impact of SARS-CoV-2 on glucose and lipid metabolism, and its emerging roles in other crucial metabolic pathways, would contribute to the occurrence and progression of COVID-19, as well as its complications and sequelae, particularly metabolic diseases.
6 Long-term effects of COVID-19The implementation of public health policies, vaccination, and comprehensive acute therapies have led to a decrease in COVID-19 mortality. However, the aftermath of this pandemic has given rise to post-acute sequelae, commonly known as long COVID or post-COVID conditions (PCC) (Brightling and Evans, 2022; Stafie et al., 2022). It has estimated that long COVID, a frequently debilitating illness, affects at least 10% of individuals surviving from COVID-19 (Davis et al., 2023). The majority of these individuals experience persistent and chronic symptoms weeks to years after the initial infection, such as fatigue, dyspnea, joint pain, chest pain, and other conditions (Lai et al., 2023). In addition, COVID-19 may often lead to more enduring and wider-ranging long-term adverse effects, including cardiovascular, cerebrovascular, metabolic, and nervous system diseases (Xie and Al-Aly, 2022; Xie et al., 2022). A recent study estimated the risks associated with 80 prespecified post-acute sequelae of COVID-19 over a period of 2 years, and found that certain post-acute sequelae, particularly those related to gastrointestinal, musculoskeletal, and neurologic systems, exhibit a prolonged risk horizon extending beyond 2 years or even longer (Bowe et al., 2023). Therefore, the knowledge on long COVID-19 may provide evidence for preventing and treating the sequelae, and offering valuable insights into its interconnectedness with other diseases.
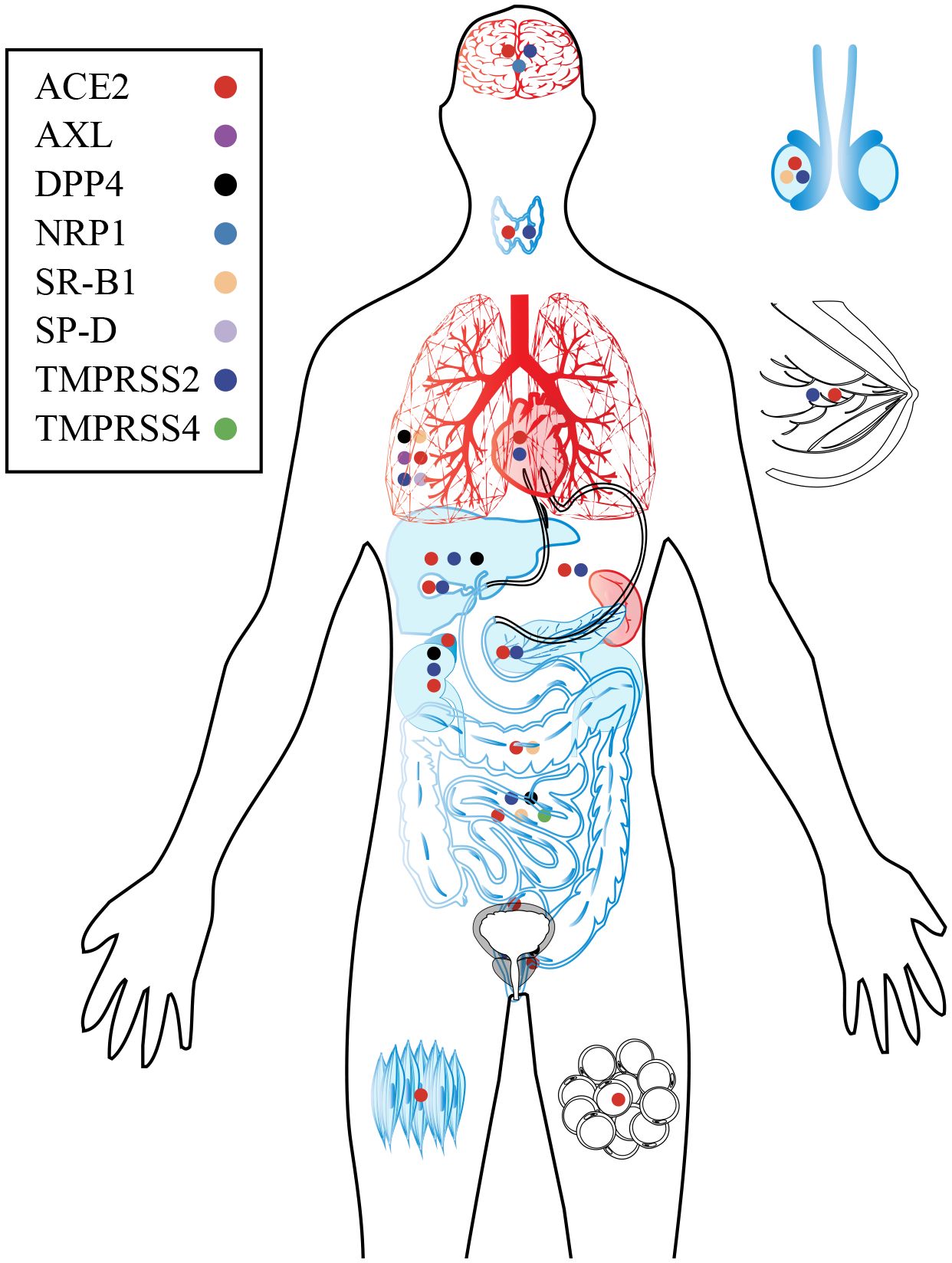
The infection of SARS-CoV-2 is contingent upon the presence of specific receptors and proteases on cell surface (Dong et al., 2020). These receptors and proteases are found in various cell types throughout the human body, including the oral and nasal mucosa, lung, heart, gastrointestinal tract, liver, kidney, spleen, brain, as well as arterial and venous endothelial cells (Pennisi et al., 2020; Sungnak et al., 2020; Nakamura et al., 2021; Qi et al., 2021; Martínez-Mármol et al., 2023), highlighting that SARS-CoV-2 may affect almost every organ, leading to acute organ damage and long-term sequelae (Figure 5). Due to the unique characteristics of various tissues and organs, the symptoms and potential long-term effects of COVID-19 vary considerably. However, major contributors are consistent and proposed to include abnormally altered immune system, circular system dysfunction, multi-organ damage, and neuroinflammation. Although mechanistic studies are generally in their early stages, it is undeniable that host factors play an indispensable role (Davis et al., 2023) (Figure 6).
Figure 5 Target organs and major receptors of SARS-CoV-2. As the primary receptor for SARS-CoV-2, ACE2 is widely distributed throughout human organs and tissues. Additionally, specific receptors in certain organs and tissues can also facilitate viral infection and subsequent damage. The organs are depicted in black, blue, and red to indicate different aspects. Black represents the distribution of SARS-CoV-2 receptors in the organ; Blue of viral infection and replication within the organ; Red indicates damage caused by the virus.
Figure 6 Long-term effects in COVID-19 survivors. By interacting with host factors, SARS-CoV-2 manipulates the intricate network of molecular mechanisms and disrupts various normal physiological mechanisms and processes in the host. Due to tissue specifi
留言 (0)