
記住我









The gut microbiota refers to the collection of various microbial communities that parasitize the host’s gastrointestinal tract, comprised of bacteria, fungi, viruses, archaea, and protozoa (Lynch and Pedersen, 2016). Typically, these microorganisms exist in a symbiotic relationship with the human host and play a crucial role in maintaining immune function and balance through interactions with the host’s immune system (La Barbera et al., 2022). The Lung-Gut axis is the concept of the mutual connection and interaction between the lungs and the gut, as these two organs are the largest surface organs with highly vascularized and immunologically active tissues. With their exposure to various external environmental challenges, including pathogenic microorganisms, harmful gases, and particulate matter, there is growing attention on the importance of the Lung-Gut Axis in impacting disease states in various ways, such as immune system communication (McAleer and Kolls, 2018), lung inflammation, and gut mucosal barrier. Changes in the gut microbiota have been linked to respiratory conditions like asthma (Barcik et al., 2020), chronic obstructive pulmonary disease (Bowerman et al., 2020), connective tissue-associated interstitial lung diseases (Salisbury et al., 2017), and acute lung injury (Kapur et al., 2018). Therefore, some gut microbial communities can serve as biomarkers for lung diseases.
Idiopathic pulmonary fibrosis (IPF) is a rare and severe chronic respiratory disease characterized by progressive interstitial lung damage and declining lung function. Symptoms of IPF include fatigue, shortness of breath during physical activity, and a persistent dry cough. This condition ultimately leads to organ failure and death. The exact cause of IPF is not fully understood, but various risk factors are thought to contribute to its development, including intrinsic factors such as genetics, aging, gender, and lung microbiota, as well as extrinsic factors such as smoking, environmental exposures, and air pollution. The incidence of IPF has been steadily increasing, likely due to factors such as aging populations and deteriorating air quality (Harari et al., 2020). IPF has an insidious onset and is often diagnosed at an advanced stage, resulting in a median survival of only 3.8 years (Neumark et al., 2020). Unfortunately, there is currently a lack of reliable diagnostic approaches in the early phase of the disease and effective treatments. Recent research suggests that dysbiosis of the gut microbiota might be associated with the progression of IPF (Ntolios et al., 2021; Quan et al., 2022). During acute exacerbation of the disease, patients often exhibit a higher microbial burden in their lungs. While there is a known correlation between the lung microbiome and disease severity, as well as the risk of disease progression and mortality, a causal relationship has yet to be established (Ntolios et al., 2021).
The link between the microbial community and IPF can be connected through related metabolites, such as fatty acids. Regarding bacterial metabolites, fatty acids may closely associate with pathophysiological processes such as mitochondrial dysfunction, functional impairment, oxidative stress, and affecting the progression of IPF (Wu et al., 2022).
Mendelian randomization (MR) analysis is a powerful tool that can leverage pre-existing aggregated data from genome-wide association studies (GWAS) to investigate the associations between complex traits and millions of molecular marker single nucleotide polymorphisms (SNPs). By using genetic variants as instrumental variables (IVs), MR can infer causality between exposure and its effect while mitigating the influence of unobserved confounding factors. Moreover, the accurate measurement of genetic variation in MR is not susceptible to measurement errors. Importantly, genetic variants are randomly allocated before birth (Birney, 2022), which aligns with the chronological order of causal timing and minimizes issues of reverse causality. As a result, MR has become an increasingly important technique in epidemiological research focusing on causal inference (de Leeuw et al., 2022). Through comparative analyses of genetic variants, MR can identify those that influence complex traits (Swerdlow et al., 2016). It offers an advantage over traditional observational studies, as it provides a robust framework for inferring causality while minimizing the risk of bias from confounding variables.
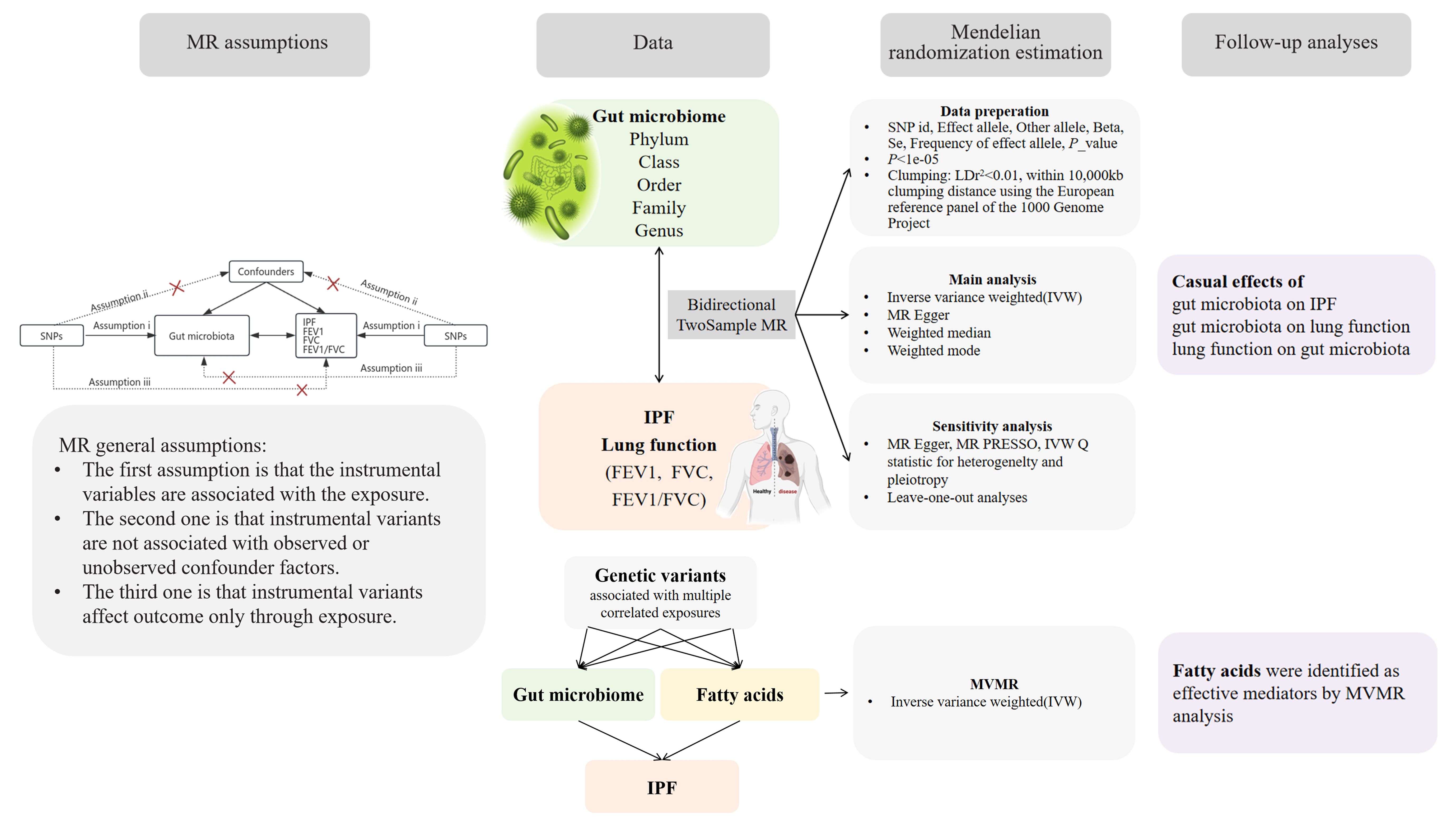
2 Materials and methods2.1 Study designFigure 1 provides an overview of the study design and assumptions underlying MR research. In MR studies, IVs must satisfy three key assumptions (Emdin et al., 2017). Assumption 1 requires that the chosen genetic variants, proposed as instrumental variables, are reliably associated with the risk factor under investigation. Assumption 2 states that the selected genetic variants should not possess any associations with potential confounding factors. Assumption 3 states that the genetic variants selected as IVs should influence the outcome risk solely through the risk factor of interest, rather than through alternative pathways.
Figure 1 A bidirectional two-sample MR model was used to evaluate the causal relationships between exposure and outcome.
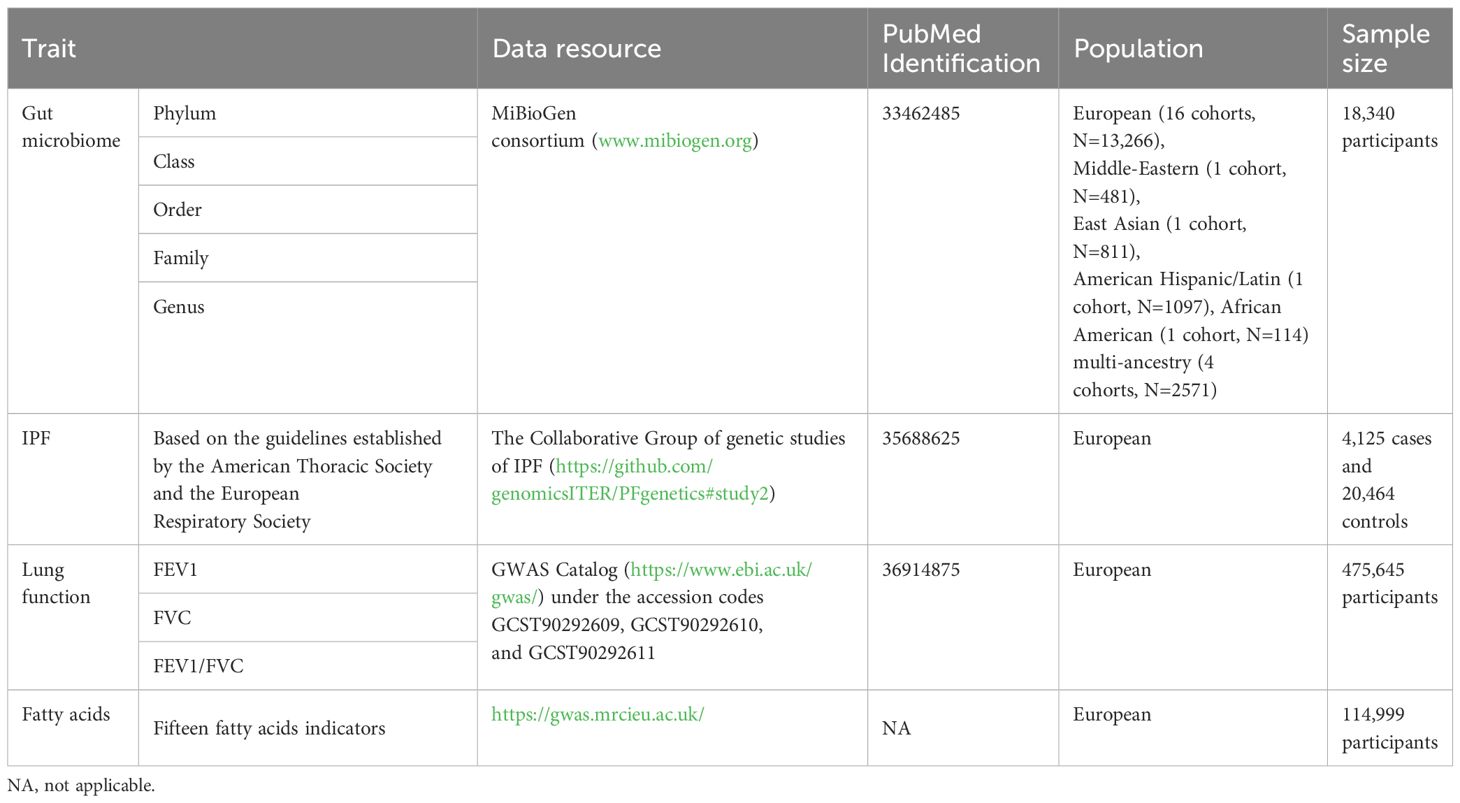
2.2 Data sources of gut microbiome and fatty acidsThe gut microbiota data used in this study were sourced from the MiBioGen consortium, which includes genome-wide genotypes and 16S fecal microbiome data from 18,340 participants across 24 cohorts, featuring 5,717,754 SNPs for a total of 211 taxa that encompass 9 phyla, 16 classes, 20 orders, 35 families, and 131 genera (Kurilshikov et al., 2021). Furthermore, we also sought to explore the potential role of fatty acids in the biological pathway of the gut microbiota to IPF. Several important fatty acids indicators were identified (including monounsaturated fatty acids, omega-3 fatty acids, omega-6 fatty acids, polyunsaturated fatty acids, ratio of docosahexaenoic acid to total fatty acids, ratio of linoleic acid to total fatty acids, ratio of monounsaturated fatty acids to total fatty acids, ratio of omega-3 fatty acids to total fatty acids, ratio of omega-6 fatty acids to omega-3 fatty acids, ratio of omega-6 fatty acids to total fatty acids, ratio of polyunsaturated fatty acids to monounsaturated fatty acids, ratio of polyunsaturated fatty acids to total fatty acids, ratio of saturated fatty acids to total fatty acids, saturated fatty acids, and total fatty acids). The GWAS data for fatty acids were extracted from MRC-IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/). Detailed information is shown in Table 1. To ensure minimal overlap with IPF studies, we specifically selected studies with no or minimal sample overlap. After calculation, we obtained a maximum overlapping rate of <10%, which may not have been sufficient to affect our results (Rees et al., 2017).
Table 1 Characteristics of the GWASs used for Analyses.
2.3 Data sources of IPF and lung functionThe data relevant to IPF were obtained from the GWAS study led by Richard J. Allen (Allen et al., 2022), which included a cohort of 4,125 cases and 20,464 controls of European ancestry from diverse regions, such as the USA, UK, and Spain. The diagnosis of IPF was conducted based on the guidelines established by the American Thoracic Society and the European Respiratory Society (Raghu et al., 2011, Raghu et al., 2018). Furthermore, the largest multi-ancestry GWAS of lung function to date, involving 475,645 participants in Europe, is accessible on the GWAS Catalog (https://www.ebi.ac.uk/gwas/) under the accession codes GCST90292609, GCST90292610, and GCST90292611 (Shrine et al., 2023).
2.4 Instrument selection and data harmonizationGiven the limited number of available SNPs, we selected SNPs significantly related to the gut microbiota with a loose cutoff of p < 1e-5. Then, significant SNPs were clumped within 10,000 kb at the level of linkage disequilibrium (LD) r2 = 0.01 using the European reference panel of the 1000 Genome Project. In reverse MR analyses, independent SNPs were selected by LD (r2 < 0.01 within 250-kb clumping distance, based on the European reference panel of the 1000 Genome Project) at a compromised significant level (1e-5) due to the relatively insufficient variables.
2.5 Statistical analysisA two-sample MR approach was implemented to investigate the causal associations between 211 microbial taxa and IPF as well as lung function. To align the effects, all SNPs were harmonized between the exposure (microbial taxa) and the outcome (IPF and lung function) based on alleles. SNPs associated with reverse causality were removed using the Steiger_filtering test (Hemani et al., 2017).
The inverse-variance weighted (IVW) method was chosen as the primary approach to estimate the total causal effect of the exposure on the outcome (Slob and Burgess, 2020). In this method, two or more IVs were combined by calculating the weighted average variance, with each IV’s weight determined as the reciprocal of the variance of the effect estimate. To complement the IVW method, we performed additional analyses including MR Egger, weighted median, and weighted mode. MR Egger allowed us to investigate the mean horizontal pleiotropic effect across instrumental variables (Burgess and Thompson, 2017). The weighted median approach generated robust estimates of the causal effect in situations where at least half the weight was derived from valid instruments, minimizing the impact of instrumental outliers (Bowden et al., 2016). Similarly, the weighted mode method assumed that the frequently observed association estimate was not influenced by pleiotropy and thus accurately reflected the true causal effect (Hartwig et al., 2017). Causal effect estimates were reported as β and Odds Ratios (OR) (OR = expβ).
We estimated the proportion of trait variance explained by the genetic instruments identified by the formula R2 = (2β2×EAF×(1-EAF))/(2β2×EAF×(1-EAF)+2N×EAF×(1-EAF)×SE2), where EAF represents the effect allele frequency, β denotes the effect size of SNP in the exposure GWAS, SE refers to the standard error, and N represents the sample size of the exposure GWAS. Instrument strength was assessed using the F statistic, where F = (R2 × (N-2))/(1-R2) (Palmer et al., 2012; Morales Berstein et al., 2022). SNPs having an F value < 10 were excluded due to their weak statistical strength. These analyses were conducted to provide a comprehensive evaluation of the causal relationships between microbial taxa and IPF, thereby enhancing the robustness and reliability of our study findings.
In an effort to identify potential vertical pleiotropic pathways that may arise from specific microbiotic metabolites, multivariable Mendelian randomization (MVMR) analyses were performed using MVMR_IVW to estimate the causal effect of specific gut microbiota on IPF after adjusting for fatty acids (Burgess and Thompson, 2015). In addition, the mr_pleiotropy_test (Verbanck et al., 2018) and IVW Q statistic (Greco et al., 2015) were utilized to identify horizontal pleiotropic outliers and quantify heterogeneity. The absence of pleiotropic effects was determined if the intercept did not significantly deviate from 0 (p>0.05). Furthermore, a leave-one-out analysis was conducted to identify potentially influential SNPs. The usage and interpretation of our MR study adhere to the STROBE-MR (Strengthening the Reporting of Observational Studies in Epidemiology-Mendelian Randomization) checklist (Skrivankova et al., 2021) (Supplementary Table S1).
All statistical analyses were undertaken using the “TwoSampleMR”, “MR-PRESSO”, and “MVMR” packages in R version 4.3.1 (http://www.r-project.org/), and a two-tailed p-value of less than 0.05 was considered statistically significant.
3 Results3.1 OverviewAfter screening for SNPs linked with exposure and removing LD, 2,875 SNPs from 211 taxa were employed as IVs. After harmonizing exposure and outcome alleles, all SNPs from various taxa performing MR analysis were shown in Supplementary Table S2. The conclusive findings between gut microbiota and IPF and lung function were summarized in Tables 2–6.
Table 2 Associations of genetic predisposition to gut microbiome with the risk of IPF.
Table 3 Associations of genetic predisposition to gut microbiome with the value of FEV1.
Table 4 Associations of genetic predisposition to gut microbiome with the value of FVC.
Table 5 Associations of genetic predisposition to gut microbiome with the value of FEV1/FVC.
Table 6 Genetic associations between lung function and gut microbiome.
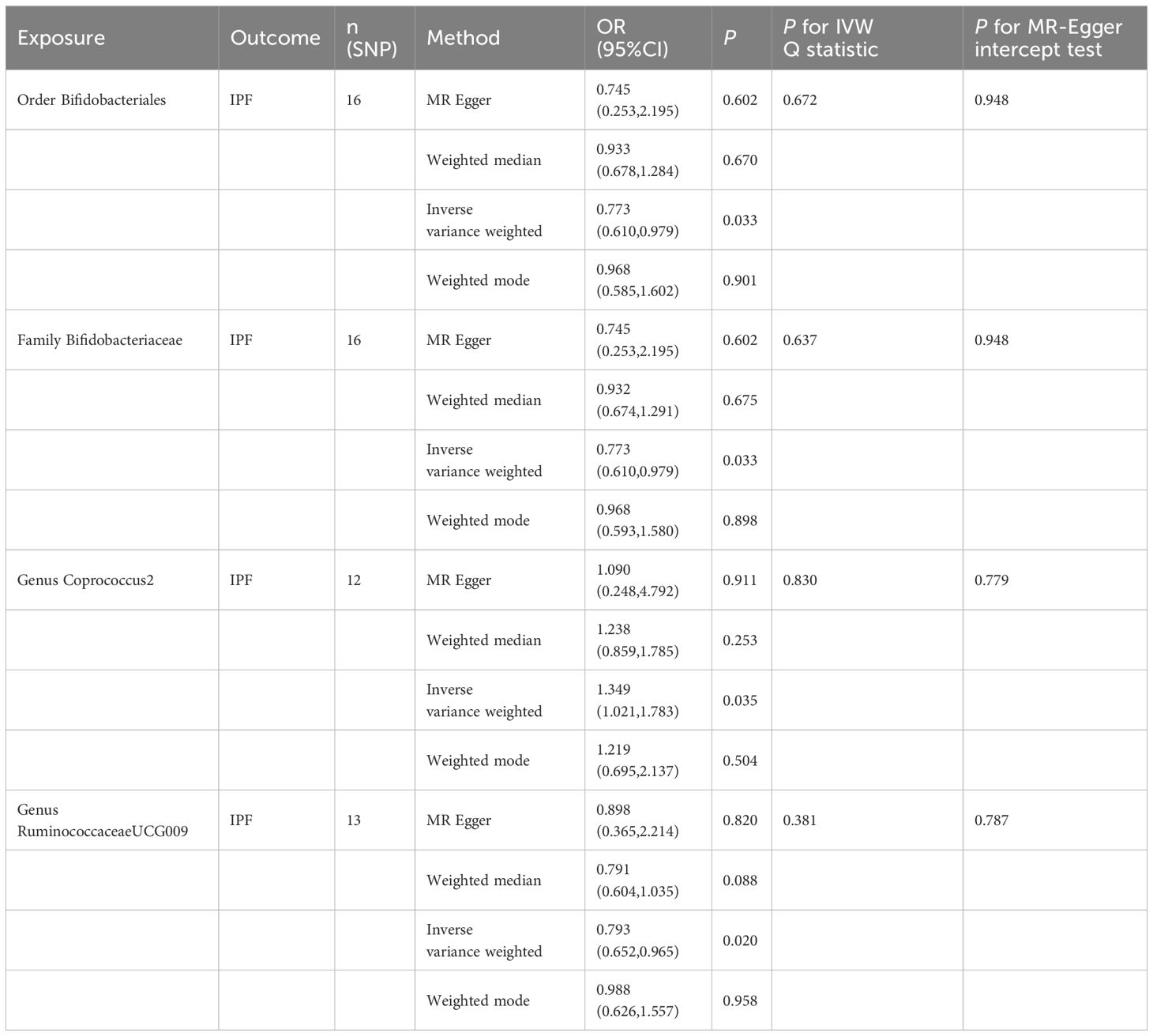
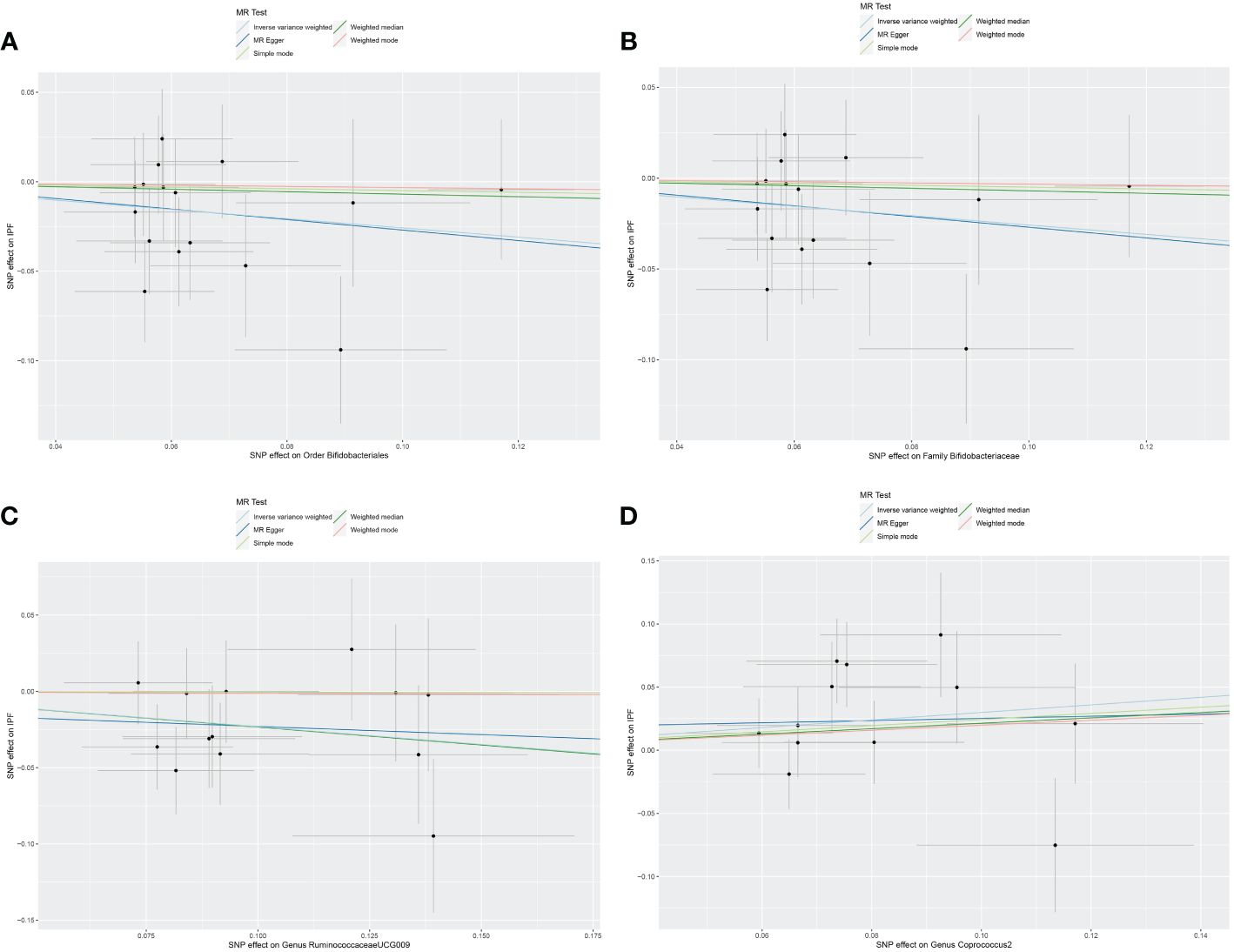
3.2 Causal effect of gut microbiota on IPFThe original GWAS involving 18,340 individuals from 24 cohorts provided summary statistics for 211 microbial taxa. Estimated by the IVW test, four taxa were identified. The MR analysis revealed that the abundance of Order Bifidobacteriales (OR=0.773, 95% CI: 0.610–0.979, p=0.033), Family Bifidobacteriaceae (OR=0.773, 95% CI: 0.610–0.979, p=0.033), and Genus RuminococcaceaeUCG009 (OR=0.793, 95% CI: 0.652–0.965, p=0.020) have a protective effect against IPF. Increased abundance of Genus Coprococcus2 was associated with a higher risk of IPF (OR=1.349, 95% CI: 1.021–1.783, p=0.035). Subsequently, Cochrane’s Q test revealed that there was heterogeneity (p<0.05). MR-PRESSO test and MR-Egger intercept tests identified no pleiotropy or significant outliers (p>0.05). The causal effect between 211 microbial taxa and IPF was presented in Table 2. The scatterplot was shown in Figure 2. All IVs used in our study were provided in Supplementary Table S2.
Figure 2 Scatter plot of the causal effect of gut microbiota on IPF. (A) Scatter plot for the causal effect of Order Bifidobacteriales on IPF risk. (B) Scatter plot for the causal effect of Family Bifidobacteriaceae on IPF risk. (C) Scatter plot for the causal effect of Genus RuminococcaceaeUCG009 on IPF risk. (D) Scatter plot for the causal effect of Genus Coprococcus2 on IPF risk.
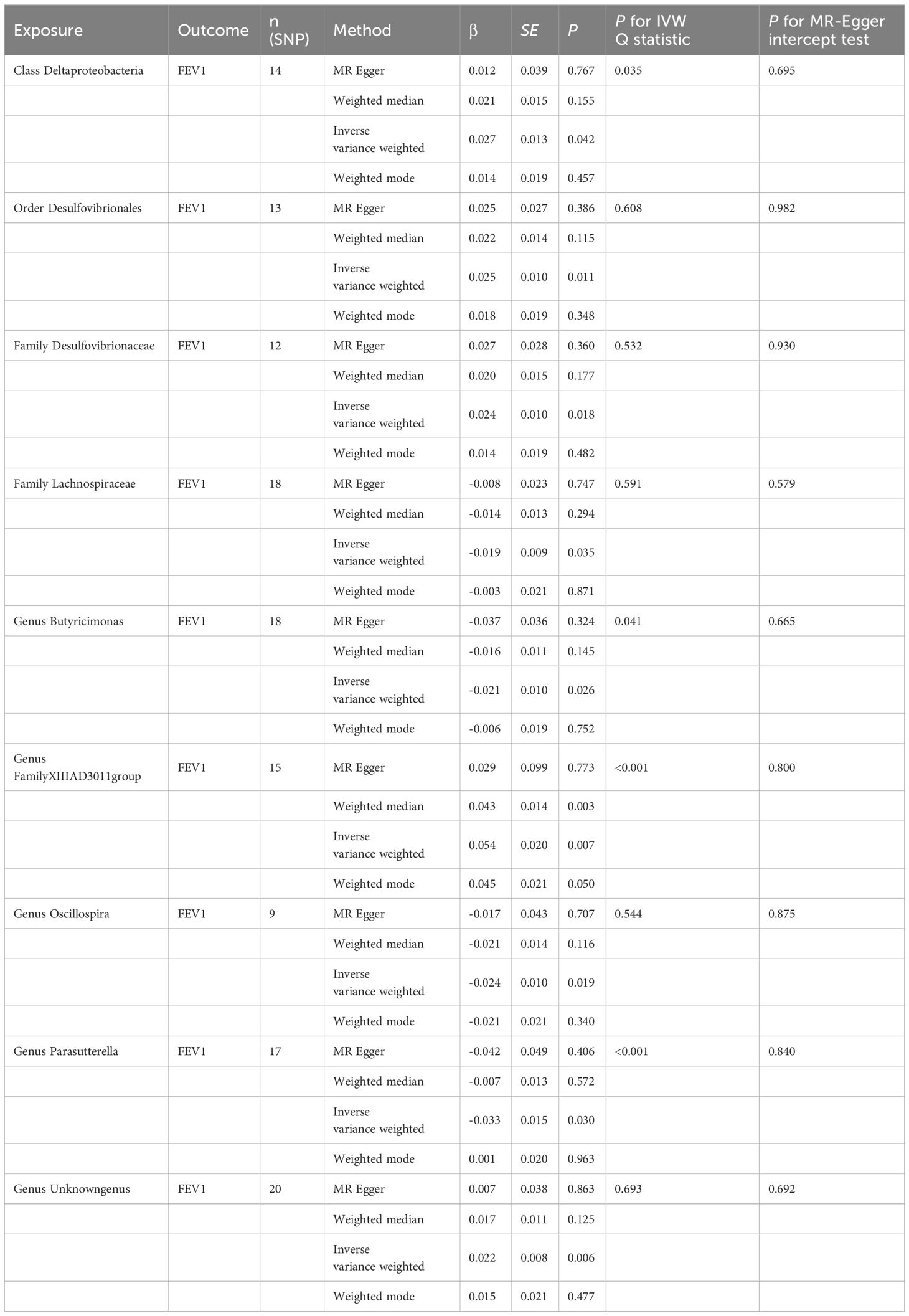
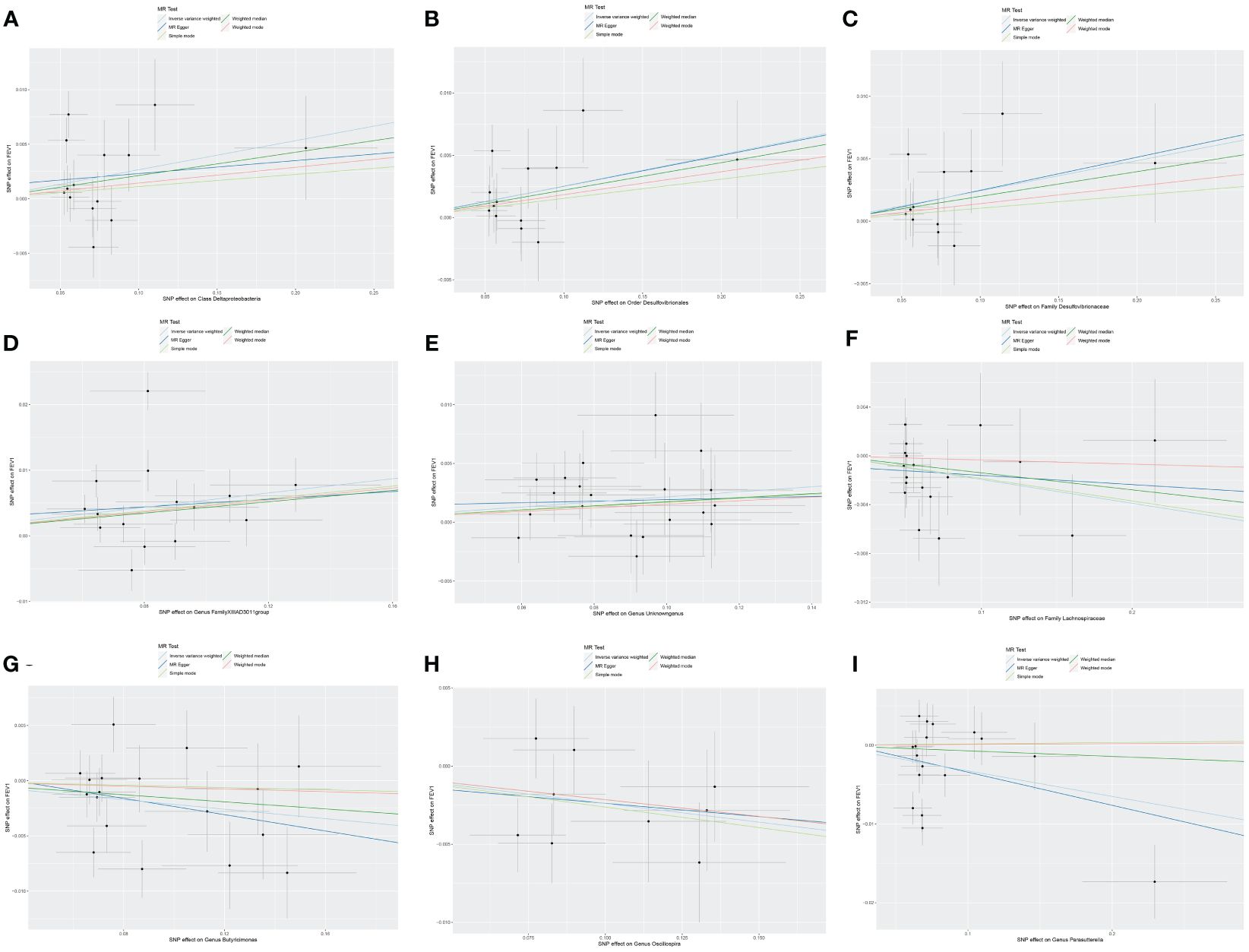
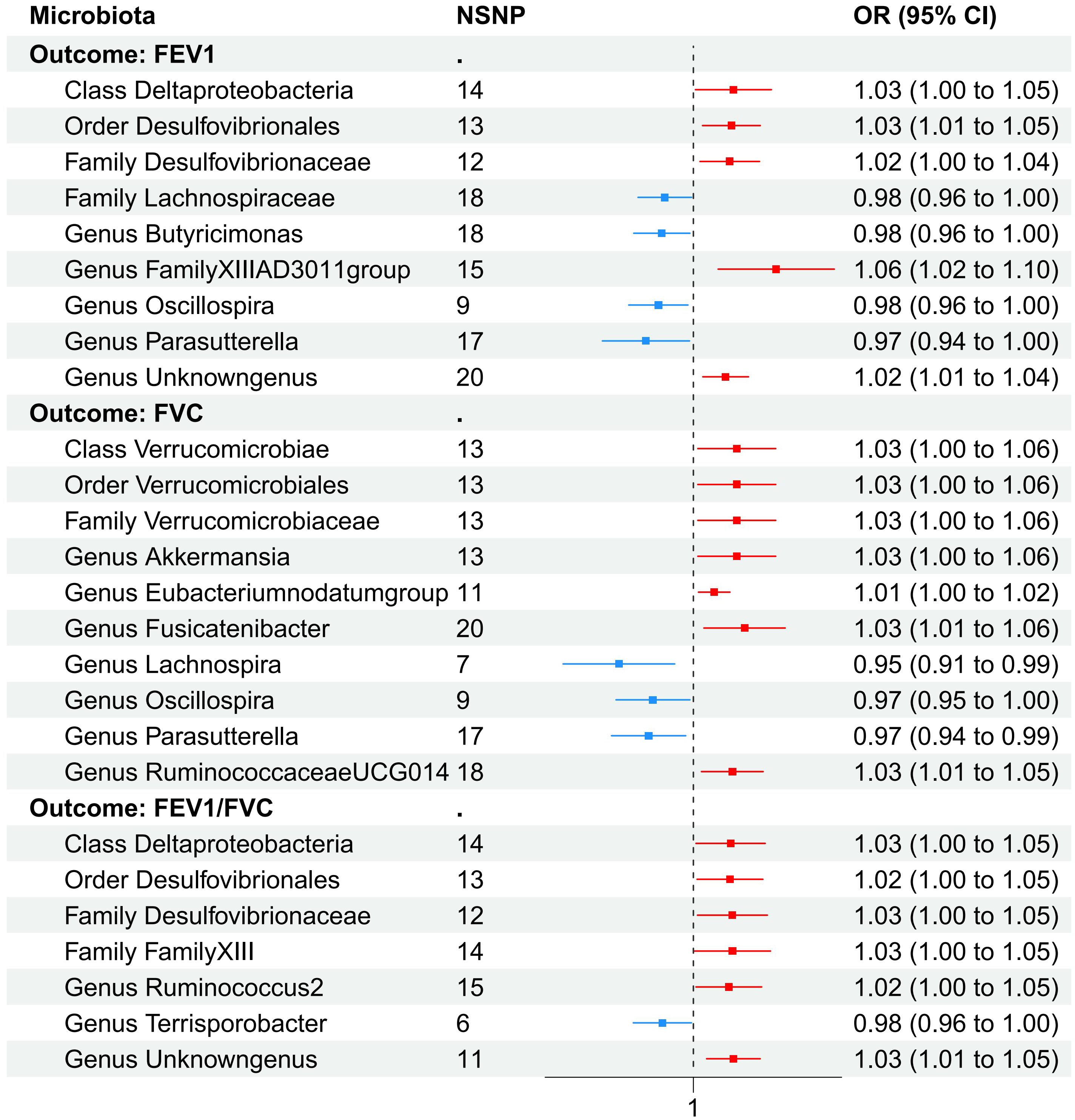
3.3 Causal effect of gut microbiota on FEV1Nine causal relationships were identified between the gut microbiota and FEV1 (Table 3). The elevation abundance of Class Deltaproteobacteria (β=0.027, se=0.013, p=0.042), Order Desulfovibrionales (β=0.025, se=0.010, p=0.011), Family Desulfovibrionaceae (β=0.024, se=0.010, p=0.018), Genus FamilyXIIIAD3011group (β=0.054, se=0.020, p=0.007), and Genus Unknowngenus (β=0.022, se=0.008, p=0.006) were associated with the raise of FEV1. However, Family Lachnospiraceae (β=-0.019, se=0.009, p=0.035), Genus Butyricimonas (β=-0.021, se=0.010, p=0.026), Genus Oscillospira (β=-0.024, se=0.010, p=0.019), and Genus Parasutterella (β=-0.033, se=0.015, p=0.030) were associated with impairment of FEV1. The results of Cochran’s Q test showed that obvious heterogeneity was found in the selected SNPs of Genus FamilyXIIIAD3011group, Genus Parasutterella, Class Deltaproteobacteria, and Genus Butyricimonas (p<0.05). No overall horizontal pleiotropy existed in any of the IVs, as shown by the results of the MR-Egger intercept test (p>0.05). Finally, the leave-one-out method achieved stable results after excluding the SNP one by one. Table 3 listed the associations of genetic predisposition to gut microbiome with the value of FEV1. The scatterplot was shown in Figure 3.
Figure 3 Scatter plot of the causal effect of gut microbiota on FEV1. (A) Scatter plot for the causal effect of Class Deltaproteobacteria on FEV1. (B) Scatter plot for the causal effect of Order Desulfovibrionales on FEV1. (C) Scatter plot for the causal effect of Family Desulfovibrionaceae on FEV1. (D) Scatter plot for the causal effect of Genus FamilyXIIIAD3011group on FEV1. (E) Scatter plot for the causal effect of Genus Unknowngenus on FEV1. (F) Scatter plot for the causal effect of Family Lachnospiraceae on FEV1. (G) Scatter plot for the causal effect of Genus Butyricimonas on FEV1. (H) Scatter plot for the causal effect of Genus Oscillospira on FEV1. (I) Scatter plot for the causal effect of Genus Parasutterella on FEV1.
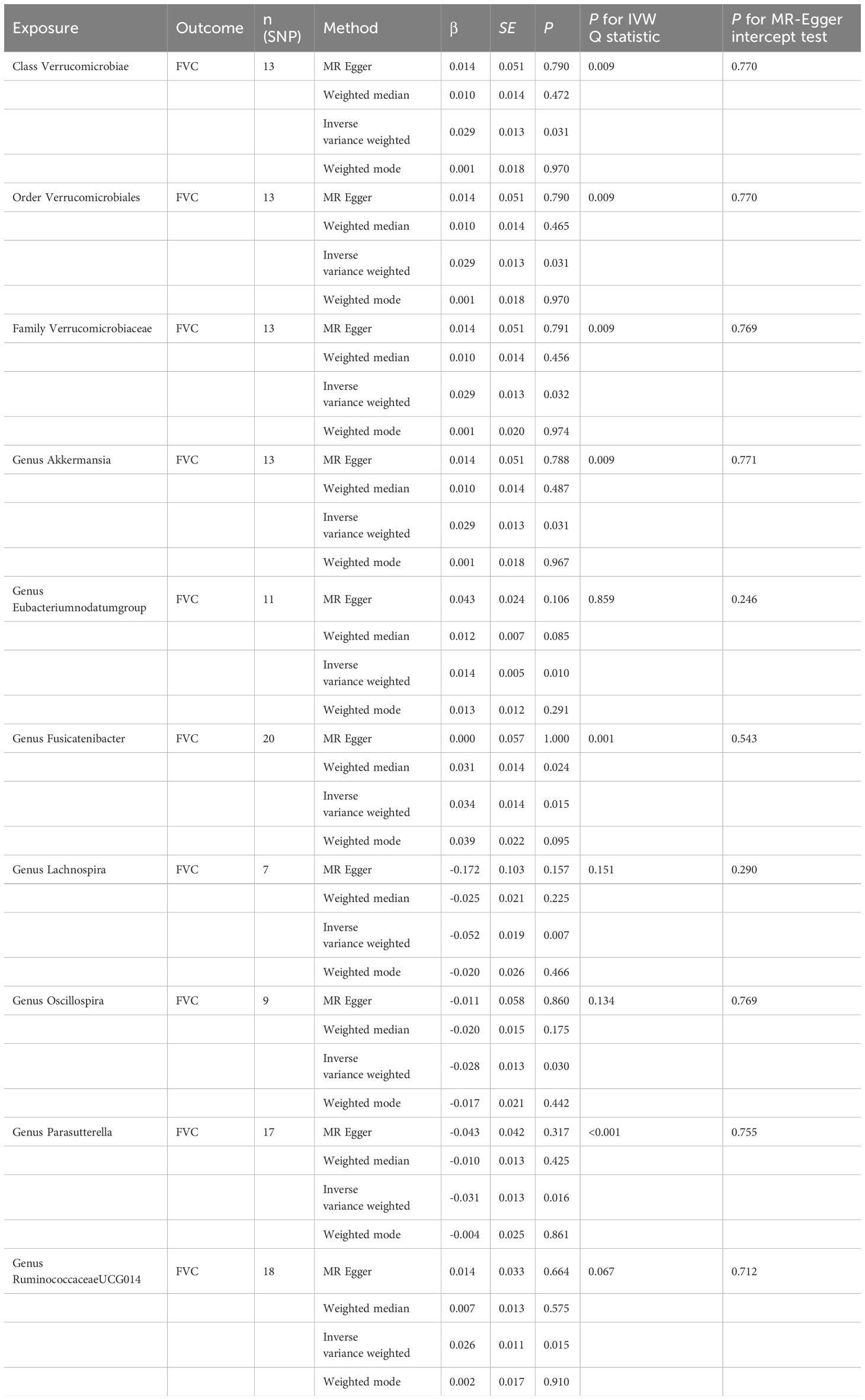
3.4 Causal effect of gut microbiota on FVCAs set out in Table 4. ten causal relationships were identified between the gut microbiota and FVC. The abundance of Class Verrucomicrobiae (β=0.029, se=0.013, p=0.031), Order Verrucomicrobiales (β=0.029, se=0.013, p=0.031), Family Verrucomicrobiaceae (β=0.029, se=0.013, p=0.031), Genus Akkermansia (β=0.029, se=0.013, p=0.031), Genus Eubacteriumnodatumgroup (β=0.014, se=0.005, p=0.010), Genus Fusicatenibacter (β=0.034, se=0.014, p=0.015), and Genus RuminococcaceaeUCG014 (β=0.026, se=0.011, p=0.015) were associated with the improvement of FVC. Genus Lachnospira (β=-0.052, se=0.019, p=0.007), Genus Oscillospira (β=-0.028, se=0.013, p=0.030), and Genus Parasutterella (β=-0.031, se=0.013, p=0.016) were associated with the reduction of FVC. To further evaluate the results, heterogeneity analyses were conducted. Results from Cochrane’s Q test showed that there was heterogeneity found in the selected SNPs among the data of Genus Akkermansia, Family Verrucomicrobiaceae, Genus Parasutterella, Class Verrucomicrobiae, Order Verrucomicrobiales, and Genus Fusicatenibacter (p<0.05). The MR-Egger intercept tests showed that there is no pleiotropy or outliers (p>0.05), suggesting that the IVs are unlikely to affect FVC through pathways other than the mentioned indicators.
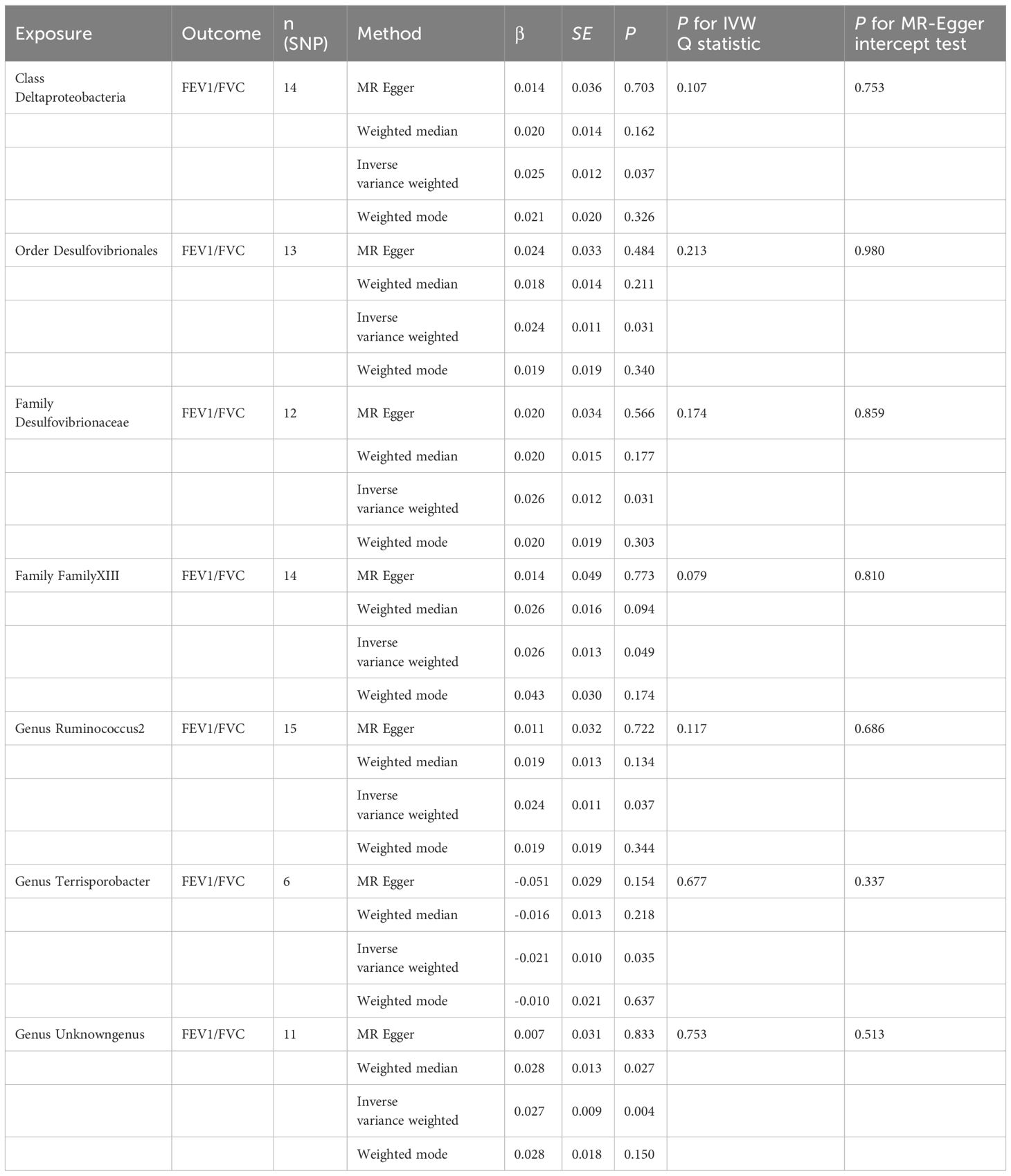
3.5 Causal effect of gut microbiota on FEV1/FVCAs shown in Table 5, Class Deltaproteobacteria (β=0.025, se=0.012, p=0.037), Order Desulfovibrionales (β=0.024, se=0.011, p=0.031), Family Desulfovibrionaceae (β=0.026, se=0.012, p=0.031), Family FamilyXIII (β=0.026, se=0.013, p=0.049), Genus Ruminococcus2 (β=0.024, se=0.011, p=0.037), and Genus unknowngenus (β=0.027, se=0.009, p=0.004) demonstrated positive associations with the increase of FEV1/FVC. Genus Terrisporobacter (β=-0.021, se=0.010, p=0.035) had a negative correlation with FEV1/FVC. Several sensitivity tests were conducted for additional confirmation of the robustness of the results. All results of Cochran’s Q test indicated that there was no significant heterogeneity (p>0.05). Moreover, the MR-Egger intercept test and the global test p-values both revealed no statistically significant results (p>0.05), suggesting no presence of horizontal pleiotropy. The leave-one-out sensitivity analysis showed stable results of the effect of Genus unknowngenus on FEV1/FVC. The forest plot illustrated all exposure factors related to pulmonary function indices (including FEV1,FVC, and FEV1/FVC) (Figure 4).
Figure 4 The forest plot illustrated all exposure factors related to pulmonary function identified by two-sample MR analysis
3.6 Causal effect of lung function on gut microbiotaTo understand the consequences of IPF and lung function on the abundance of the gut microbiome, reverse two-sample MR tests were performed. Due to the insufficient number of analyzable SNPs when considering IPF and FEV1/FVC as exposure factors, further MR analysis and subsequent heterogeneity analyses cannot be conducted. Therefore, the reverse MR analysis in this study only focuses on the casual effects of FEV1 and FVC on the abundance of gut microbiota.
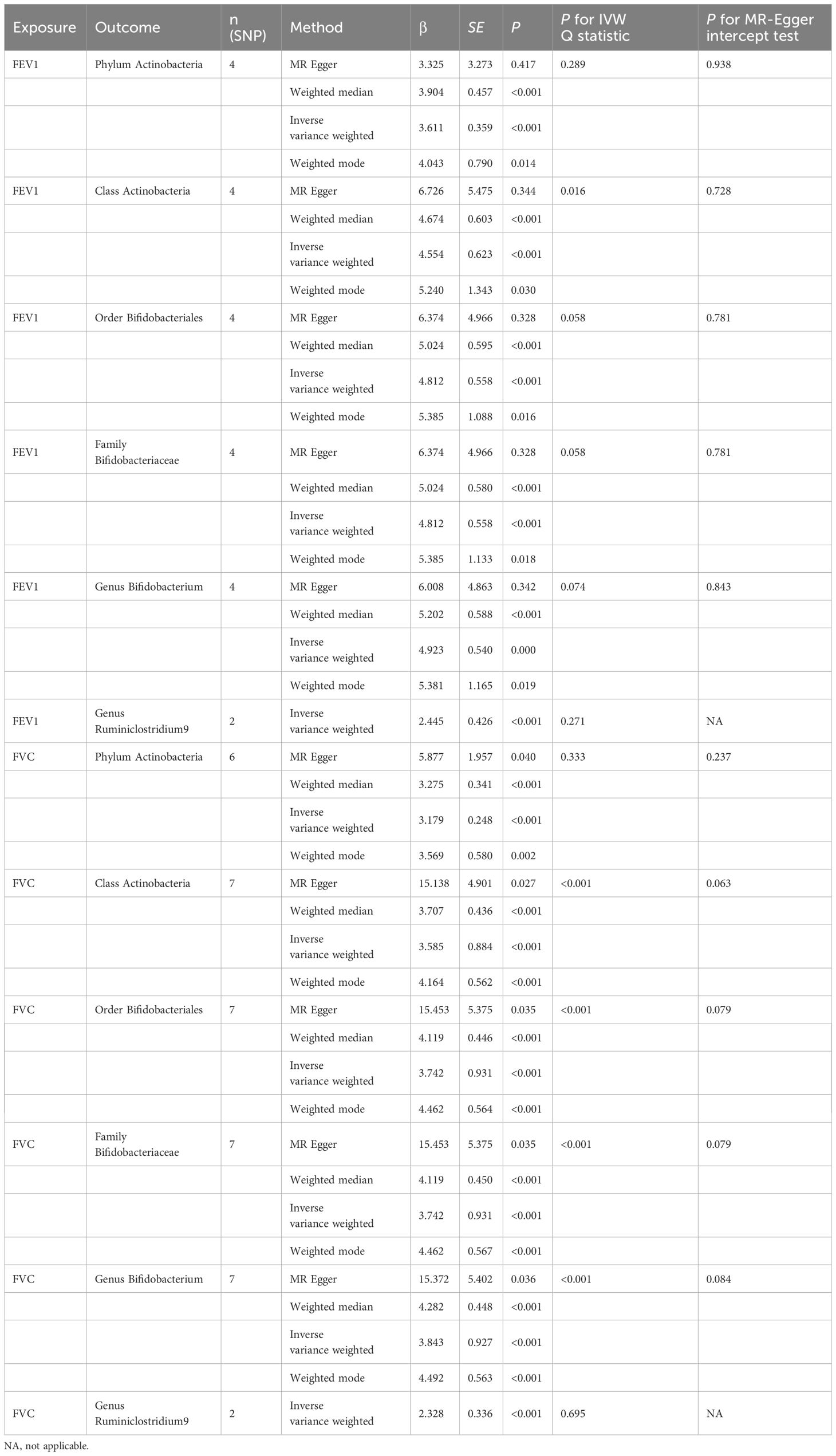
The increase of FEV1 can amplify the abundance of Phylum Actinobacteria (β=3.611, se=0.359, p<0.001), Class Actinobacteria (β=4.554, se=0.623 p<0.001), Order Bifidobacteriales (β=4.812, se=0.558, p<0.001), Family Bifidobacteriaceae (β=4.812, se=0.558, p<0.001), Genus Bifidobacterium (β=4.923, se=0.540, p<0.001), and Genus Ruminiclostridium9 (β=2.445, se=0.426, p<0.001). When investigating the impact of FVC on gut microbiota, a promoting trend for the abundance of the same gut microbiota was observed. Detailed significant results for the causal relationships are listed in Table 6. None horizontal pleiotropy was detected at statistically significant levels (all p for MR-Egger intercept test > 0.05).
3.7 Fatty acids were identified as effective mediators by MVMR analysisConsidering the possible impact of fatty acids on the progression from specific gut microbiota to IPF, MVMR analysis was conducted by adjusting for significant associations with fifteen indicators of fatty acids. Drawing on earlier research findings of this study, we hypothesized that the enhancement of FEV1 could lead to increased abundance of Bifidobacteriales, resulting in a reduced risk of IPF.
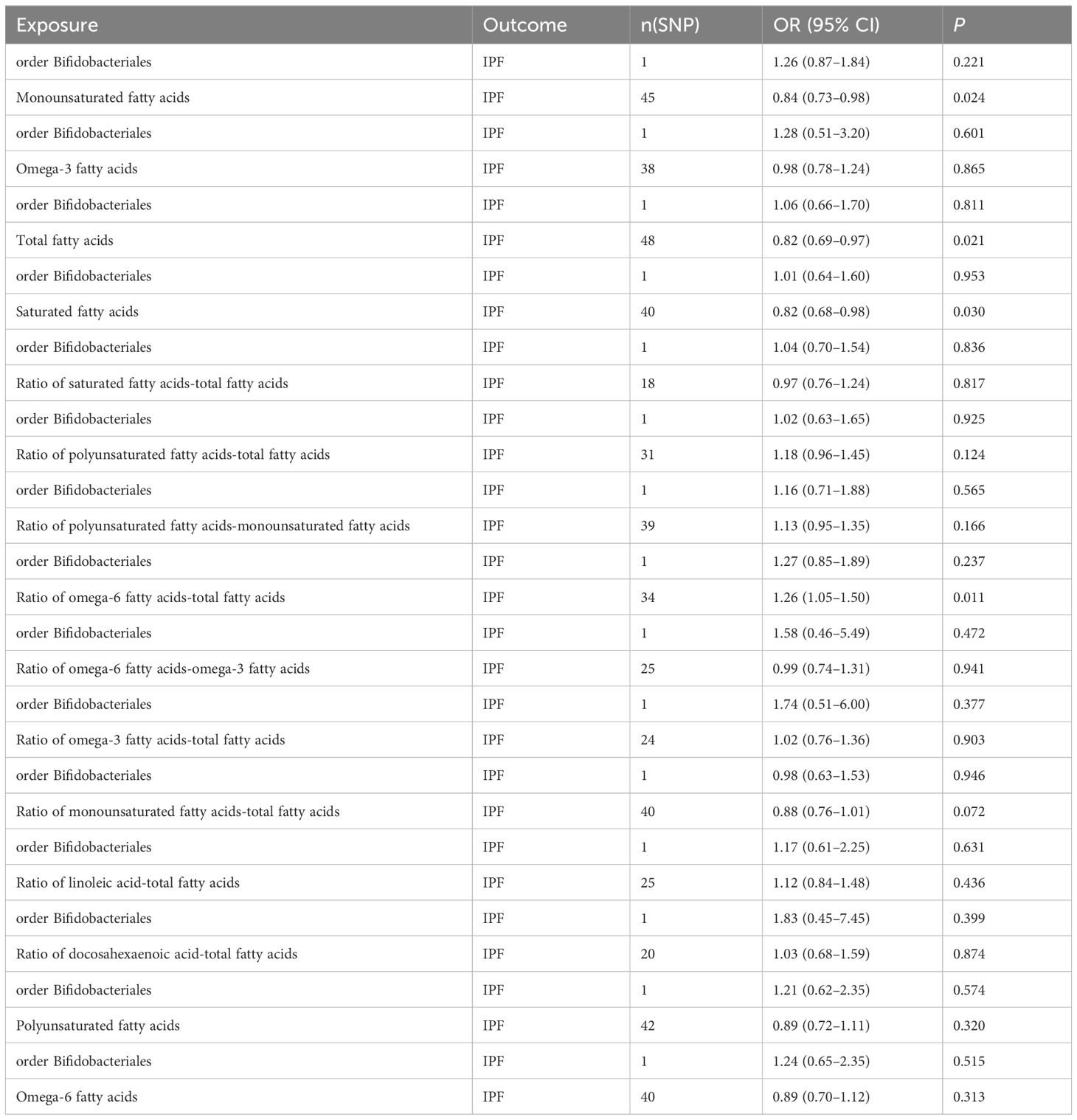
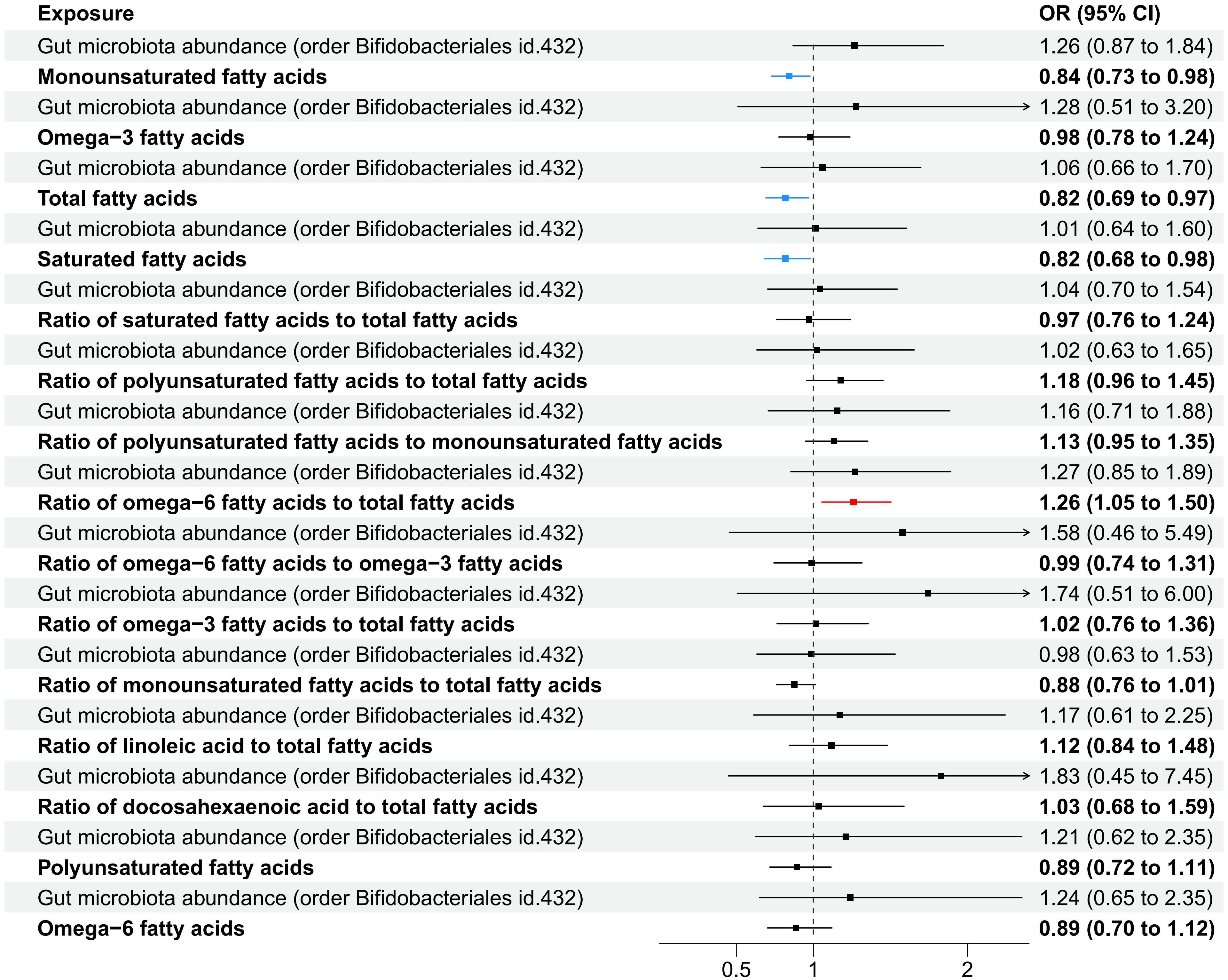
In the MVMR analysis, our focus was on the abundance of gut microbiota of the Bifidobacteriales order. Adjustments were made for one fatty acid at a time, with Bifidobacteriales serving as a co-exposure, to assess their potential mediation effects on IPF. The findings, as presented in Table 7. Four factors related to fatty acids showed potential mediation in linking Bifidobacteriales and IPF: monounsaturated fatty acids (OR=0.84, 95% CI: 0.73–0.98, p=0.024), total fatty acids (OR=0.82, 95% CI: 0.69–0.97, p=0.021), saturated fatty acids (OR=0.82, 95% CI: 0.68–0.98, p=0.030), and the ratio of omega-6 fatty acids to total fatty acids (OR=1.26, 95% CI: 1.05–1.50, p=0.011). The forest plot depicting these results is presented in Figure 5.
Table 7 MVMR results of causal relationships between gut microbiota abundance (order Bifidobacteriales) and IPF after adjusting for fatty acids.
Figure 5 A forest plot displayed the factors related to fatty acids identified through MVMR analysis, with Order Bifidobacteriales as the exposure and IPF as the outcome variable.
4 DiscussionUsing the summary statistics of gut microbiota from the largest GWAS meta-analysis conducted by the MiBioGen consortium (Kurilshikov et al., 2021) and the latest summary statistics of IPF (Allen et al., 2022), FEV1, FVC and FEV1/FVC (Shrine et al., 2023) limited to European ancestry, we performed a bidirectional two-sample MR analysis to evaluate the causal association between gut microbiota and IPF and lung function. Four taxa were found causally associated with the risk of IPF. Order Bifidobacteriales, Family Bifidobacteriaceae, and Genus RuminococcaceaeUCG009 exerted protective effects on IPF, while Genus Coprococcus2 promote the development of IPF. Several taxa were causally associated with lung function. Among them, the most prominent beneficial microbiota comprised by Class Deltaproteobacteria, Order Desulfovibrionales, Family Desulfovibrionaceae, Class Verrucomicrobiae, Order Verrucomicrobiales, and Family Verrucomicrobiaceae. Meanwhile, Family Lachnospiraceae, Genus Oscillospira, and Genus Parasutterella were associated with the impairment of lung function. In the reverse MR analysis, the abundance of Order Bifidobacteriales, Family Bifidobacteriaceae, and Genus Bifidobacterium increased with the improvement of FEV1 and FVC. The MVMR results suggested that fatty acids (monounsaturated fatty acids, total fatty acids, saturated fatty acids, and ratio of omega-6 fatty acids to total fatty acids) probably played a role in the genetic pathway from the gut microbiota to IPF, especially for Bifidobacteriales.
The lung is the largest human organ in direct contact with the environment. The lung was considered non-sterile partly due to the failure of isolating bacteria in lung specimens with traditional culture techniques. Only in the setting of infections, such as pneumonia or bronchiectasis, as in such disorders microbes could be culture-isolated and considered pathogenic of the disease (Ntolios et al., 2021). The introduction of sequencing of the 16S rRNA gene technique allowed recognition of the fact that bacteria not only exist within the human lung but are altered in lung disease and correlate with alveolar immunity and clinical outcomes (Invernizzi et al., 2020). The normal lung microbiota is primarily composed of gram-negative bacteria and facultative anaerobes. A previous MR analysis has suggested that gut microbiota can impact chronic respiratory diseases (CRDs), including chronic obstructive pulmonary disease (COPD), asthma, interstitial lung disease (ILD), sarcoidosis and occupational lung diseases (Shi et al., 2023). The presence and abundance of specific gut microbiota in various disease conditions imply a potential role in modulating immune responses and contributing to the development or resolution of inflammation-related disorders.
Utilizing 16S rRNA gene sequencing, Wei’s research (Wei et al., 2023) indicates that bleomycin (BLM) induced pulmonary fibrosis (PF) could alter the relative abundance of many microbiotas in mice gut. At the family level, compared to the control group, the abundance of Bifidobacteriaceae, Erysipelotrichaceae, and Lactobacillaceae showed an elevation in PF group, while some beneficial microbiota was significantly decreased, such as Bacilaceae and Lachnospiraceae. At the genus level, the abundance of Akkermansia, Bacillus, and Lactobacillus showed reduction in the PF group while was significantly increased for Clostridium, Erysipelatoclostridium, Faecalibaculum, and Lachnoclostridium compared to the control group. Consistent with Wei’s study, we used MR to validate the genetic beneficial effect of Genus Akkermansia on lung function. Recent investigations have elucidated the significance of Akkermansia muciniphila (A. muciniphila) in modulating the pathophysiology of interstitial lung diseases, notably in cystic fibrosis (CF) and COVID-19 convalescent patients. Pharmacological interventions aimed at augmenting A. muciniphila populations have demonstrated the potential to alleviate intestinal inflammation (Manor et al., 2016), diminish pathogenic bacterial loads, and thereby emerge as a therapeutic target for microbiome-based therapies in CF. According to a study performed on SARS-CoV-2 recovered patients (Yeoh et al., 2021), an elevated presence of A. muciniphila correlates with markers of inflammation, implicating its association with dysbiosis of the gut microbiota and systemic inflammatory responses during disease states. Yoon and colleagues (Yoon et al., 2021) extracted genomic DNA from lung tissues of patients with IPF and found that the relative abundance of Lactobacillus, Paracoccus, and Akkermansia was increased in patients with IPF compared with that in the controls. Based on the findings of our study, which suggest that Akkermansia acts as a protective factor for lung function, it is warranted for further investigation of the specific mechanisms of Akkermansia for the impact on IPF. Wei et.al (Wei et al., 2023) also concluded that Family Bifidobacteriaceae would exacerbate the microbial burden in IPF and promote disease progression, while our findings suggested that an increase in FEV1 can give rise to the abundance of Family Bifidobacteriaceae. The latter was present as a probiotic to reduce the risk of IPF.A study (Yeoh et al., 2021) conducted on individuals who recovered from SARS-CoV-2 also observed that certain species of Bifidobacteria were present at reduced levels.
Quan and colleagues (Quan et al., 2022) showed that after BLM induced PF in mice, the microecological balance of the gut microbiota was destroyed, and the relative abundance of some intestinal probiotics like Firmicutes, Lactobacillales, Lactobacillaceae, Lactobacillus, and Catenibacterium dramatically lowered while the relative abundance of Verrucomicrobiales and Enterobacteriales remarkably increased. Differently, our study found that Family Lachnospiraceae was associated with impairment of FEV1. Yoon’s study (Yoon et al., 2021) revealed a relatively increased abundance of Lactobacillus and Bifidobacterium in the lung tissue of IPF patients. Lactobacillus generally resides in the gastrointestinal and reproductive tract, where it maintains a healthy microecology with lactic acid production. However, given the well-known association between IPF and gastroesophageal reflux disease (Cheng et al., 2023), the high prevalence of GERD in IPF might contribute to the increase in the relative abundance of Lactobacillus in IPF. Levels of lactic acid and lactate dehydrogenase-5, which induce the differentiation of fibroblasts into myofibroblasts by activating transforming growth factor (TGF)-ß1, were elevated in lung tissues from patients with IPF compared with healthy persons (Kottmann et al., 2012). Therefore, bacteria that produce lactic acid might also contribute to the progression of IPF. However, a different conclusion drawn by wang and colleagues (Wang et al., 2022) was that Lactobacillus mucosae can regulate immune responses and intestinal micro-ecological balance by reducing the proportions of inflammatory cells, including granulocytes and monocytes in the blood, and increasing interferon (IFN)-β, interleukin (IL)-1β, IL-10, and tumor necrosis factor (TNF)-α levels. The observed discrepancies in the role of lactic acid-producing bacteria, notably Lactobacillus mucosae, concerning IPF progression, may be attributed to multifactorial influences and intricate mechanisms. Firstly, strain-specific variations play a pivotal role, as different strains within the same species exhibit diverse immunomodulatory capacities, thereby influencing their impact on IPF. Secondly, individual host characteristics, encompassing genetic predispositions, immune status, and general health conditions, exert a substantial effect on the host’s response to bacterial interventions. This inherent variability among IPF patients might account for the divergent outcomes following exposure to Lactobacillus mucosae. Lastly, the efficacy of Lactobacillus mucosae seems to be intricately tied to the administered dose and duration of treatment, thereby accentuating the necessity for meticulous attention to these variables in forthcoming research endeavors.
This MR study found that an increase in Genus Parasutterella abundance leads to a decrease in FEV1 and FVC. Gong and colleagues (Gong et al., 2021) calculated the sequence proportions of microbiome and made comparison between fibrotic animals and control ones, showing that Parasutterella were synchronously up-regulated in PF group and Parasutterella was negatively correlated with thymidine. There are very close correlations between distinctive gut microbiota and metabolites under pulmonary fibrotic pathological conditions (Bai et al., 2019; Fang et al., 2020). In recent scientific explorations centered around the “gut-lung axis,” alterations in gut microbial composition and their metabolic byproducts have been implicated in the development of fibrotic interstitial lung diseases. The gut microbiome participates in lung fibrogenesis through metabolically mediated pathways. TGF-β-driven stimulation of fibroblasts enhances glutamine and glutamate concentrations, necessitating glutaminolysis for myofibroblast differentiation and activation (Bernard et al., 2018). In macrophages, arginine boosts glutathione levels and curbs the secretion of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, with arginine derivatives like iNOS and cNOS exerting bidirectional effects on airway inflammation (Fu et al., 2020; Wu et al., 2021). Arginine also restrains NF-κB activation and suppresses MMP-2 and MMP-9 activities implicated in fibrosis (Hnia et al., 2008). Tryptophan, interacting with aryl hydrocarbon receptors, dampens pro-inflammatory T-cell subsets (Takei et al., 2020), and upon conversion to 5-MTP in fibroblasts, it impedes macrophage activation and inflammatory mediator release by interfering with TGF-β/SMAD3 and PI3K/Akt signaling, concurrently hindering myofibroblast formation (Fang et al., 2020). Furthermore, butyrate inhibits TGF-β and fibroblast expression through HDAC-mediated histone acetylation (Park et al., 2021), whereas bile acids stimulate the TGF-β1/Smad3 pathway, promoting alveolar epithelial and lung fibroblast activation (Chen et al., 2017). Collectively, these findings underscore the complex interplay between gut-derived metabolites and the progression of lung fibrosis, highlighting potential therapeutic avenues.
Our study suggested the potential protective roles of specific monounsaturated fatty acids and saturated fatty acids in the genetic association between Bifidobacterium and IPF. Previous research demonstrated that exposure to a high-fat diet rich in palmitic acid, a saturated fatty acid, increased lung fibrosis in wild-type mice after being administered bleomycin. This effect was found to be associated with the activation of the unfolded protein response and apoptosis of lung epithelial cells (Chu et al., 2019), suggesting that high intake of saturated fatty acids may be a contributing factor to the pathogenesis of EMT due to a defect in long-chain fatty acid family member 6 enzyme. Additionally, intratracheal administration of fatty acids has shown potential for therapeutic application in the treatment of PF (Zhao et al., 2014). These findings suggest that a diet rich in essential fatty acids or targeted delivery of fatty acids directly to the lungs may represent a promising approach for the prevention and treatment of lung fibrosis.
To our knowledge, this study is the first comprehensive investigation utilizing large-scale MR analysis to examine the causal relationship between gut microbiota, fatty acids, IPF, and lung function. We utilized the latest and largest GWAS data of individuals with European ancestry. The strength of the study lies in the use of MR analysis, which reduces the impact of measurement errors and addresses potential issues such as reverse causation and confounding factors commonly associated with observational studies. Furthermore, we conducted various sensitivity analyses using multiple complementary MR approaches to assess the robustness of the association and potential bias from pleiotropy. Overall, this study provides valuable insights into the gut-lung axis in IPF and contributes to the understanding of potential preventive and treatment strategies for this condition.
This study has limitations that need to be acknowledged. First, the study population is limited to individuals of European ancestry, thereby rendering the findings not generalizable to populations of other ancestral backgrounds. As IPF prevalence varies across regions and races, further research is warranted to determine regional or racial disparities. Second, heterogeneity among IPF patients may lead to inconsistent results, limiting the conclusions that can be drawn. The underlying mechanism of the lung-gut axis in IPF remains unclear as our research focuses on correlation analysis. Therefore, further investigations are needed to explore specific mechanisms. Comprehensive studies on the lung-gut axis are essential as factors such as seasonality, age, living habits, diet structure, genetic background, and treatment regimens may affect fecal composition in IPF patients. Such studies may provide new directions and strategies for the prevention and treatment of IPF.
5 ConclusionIn summary, the current study suggested the casual effects of the specific gut microbes on the risk of IPF and lung function. In turn, lung function also exerted a positive role in some gut microbes. Maintaining an appropriate dietary consumption of lipid substances can provide a certain level of protection against the development and progression of IPF. Our findings provided novel insights into the potential role of gut microbiota for IPF and indicated a potential mechanism of gut microbiota-mediated prevention of IPF.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributionsYR: Writing – original draft, Software, Methodology, Investigation, Formal Analysis, Data curation, Conceptualization. YZ: Writing – review & editing, Methodology, Investigation, Data curation, Conceptualization. YC: Writing – review & editing, Methodology, Investigation, Data curation, Conceptualization. HQ: Writing – review & editing, Methodology, Investigation, Data curation, Conceptualization. HZ: Writing – review & editing, Supervision, Project administration, Funding acquisition.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Shanxi Province science and technology cooperation and exchange special project (Regional cooperation project) (202204041101031), a Research Project Supported by the Shanxi Scholarship Council of China (2023-190), and Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province ((2014) 779).
AcknowledgmentsThe authors would like to thank the networks for providing the data by MiBioGen consortium (www.mibiogen.org), MRC-IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/), the Collaborative Group of genetic studies of IPF (https://github.com/genomicsITER/PFgenetics#study2), and GWAS Catalog (https://www.ebi.ac.uk/gwas/).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1348685/full#supplementary-material
AbbreviationsMR, Mendelian randomization; GWAS, Genome-wide association studies; SNPs, Single nucleotide polymorphisms; IPF, Idiopathic pulmonary fibrosis; IVs, Instrumental variables; IVW, Inverse-variance weighted; OR, Odds ratio; CI, Confidence interval; FEV1, Forced expiratory volume in one second; FVC, Forced vital capacity; FEV1/FVC, Forced expiratory volume in one second/forced vital capacity.
ReferencesAllen, R. J., Stockwell, A., Oldham, J. M., Guillen-Guio, B., Schwartz, D. A., Maher, T. M., et al. (2022). Genome-wide association study across five cohorts identifies five novel loci associated with idiopathic pulmonary fibrosis. Thorax 77, 829–833. doi: 10.1136/thoraxjnl-2021-218577
PubMed Abstract | CrossRef Full Text | Google Scholar
Bai, L., Bernard, K., Tang, X., Hu, M., Horowitz, J. C., Thannickal, V. J., et al. (2019). Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 60, 49–57. doi: 10.1165/rcmb.2018-0180OC
PubMed Abstract | CrossRef Full Text | Google Scholar
Barcik, W., Boutin, R. C. T., Sokolowska, M., Finlay, B. B. (2020). The role of lung and gut microbiota in the pathology of asthma. Immunity 52, 241–255. doi: 10.1016/j.immuni.2020.01.007
PubMed Abstract | CrossRef Full Text | Google Scholar
Bernard, K., Logsdon, N. J., Benavides, G. A., Sanders, Y., Zhang, J., Darley-Usmar, V. M., et al. (2018). Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J. Biol. Chem. 293, 1218–1228. doi: 10.1074/jbc.RA117.000444
PubMed Abstract | CrossRef Full Text | Google Scholar
Bowden, J., Davey Smith, G., Haycock, P. C., Burgess, S. (2016). Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314. doi: 10.1002/gepi.21965
PubMed Abstract | CrossRef Full Text | Google Scholar
Bowerman, K. L., Rehman, S. F., Vaughan, A., Lachner, N., Budden, K. F., Kim, R. Y., et al. (2020). Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 11, 5886. doi: 10.1038/s41467-020-19701-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Burgess, S., Thompson, S. G. (2015). Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 181, 251–260. doi: 10.1093/aje/kwu283
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, B., You, W. J., Liu, X. Q., Xue, S., Qin, H., Jiang, H. D. (2017). Chronic microaspiration of bile acids induces lung fibrosis through multiple mechanisms in rats. Clin. Sci. (Lond) 131, 951–963. doi: 10.1042/CS20160926
PubMed Abstract | CrossRef Full Text | Google Scholar
Cheng, X., Shi, J., Zhang, D., Li, C., Xu, H., He, J., et al. (2023). Assessing the genetic relationship between gastroesophageal reflux disease and chronic respiratory diseases: a mendelian randomization study. BMC Pulm Med. 23, 243. doi: 10.1186/s12890-023-02502-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Chu, S. G., Villalba, J. A., Liang, X., Xiong, K., Tsoyi, K., Ith, B., et al. (2019). Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 61, 737–746. doi: 10.1165/rcmb.2018-0324OC
PubMed Abstract | CrossRef Full Text | Google Scholar
de Leeuw, C., Savage, J., Bucur, I. G., Heskes, T., Posthuma, D. (2022). Understanding the assumptions underlying Mendelian randomization. Eur. J. Hum. Genet. 30, 653–660. doi: 10.1038/s41431-022-01038-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Fang, L., Chen, H., Kong, R., Que, J. (2020). Endogenous tryptophan metabolite 5-Methoxytryptophan inhibits pulmonary fibrosis by downregulating the TGF-β/SMAD3 and PI3K/AKT signaling pathway. Life Sci. 260, 118399. doi: 10.1016/j.lfs.2020.118399
PubMed Abstract | CrossRef Full Text | Google Scholar
Fu, A., Alvarez-Perez, J. C., Avizonis, D., Kin, T., Ficarro, S. B., Choi, D. W., et al. (2020). Glucose-dependent partitioning of arginine to the urea cycle protects β-cells from inflammation. Nat. Metab. 2, 432–446. doi: 10.1038/s42255-020-0199-4
PubMed Abstract | CrossRef Full Text | Google Scholar
Gong, G. C., Song, S. R., Su, J. (2021). Pulmonary fibrosis alters gut microbiota and associated metabolites in mice: An integrated 16S and metabolomics analysis. Life Sci. 264, 118616. doi: 10.1016/j.lfs.2020.118616
PubMed Abstract | CrossRef Full Text | Google Scholar
Greco, M. F., Minelli, C., Sheehan, N. A., Thompson, J. R. (2015). Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 34, 2926–2940. doi: 10.1002/sim.6522
PubMed Abstract | CrossRef Full Text | Google Scholar
Harari, S., Davì, M., Biffi, A., Caminati, A., Ghirardini, A., Lovato, V., et al. (2020). Epidemiology of idiopathic pulmonary fibrosis: a population-based study in primary care. Intern. Emerg. Med. 15, 437–445. doi: 10.1007/s11739-019-02195-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Hartwig, F. P., Davey Smith, G., Bowden, J. (2017). Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998. doi: 10.1093/ije/dyx102
PubMed Abstract | CrossRef Full Text | Google Scholar
Hemani, G., Tilling, K., Davey Smith, G. (2017). Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PloS Genet. 13, e1007081. doi: 10.1371/journal.pgen.1007081
留言 (0)