Animals and treatment
The 5-week-old male C57BL/6J mice used in this study were purchased from GemPharmatech. For the HFD mouse model, the mice were randomly divided into an HFD group and an ND group, with 6 mice in each group. Starting from the 5th week of age, the regular feed of mice in the HFD group was replaced with an HFD (D12492; Research Diet), while the ND group mice were consistently fed an ND until they were euthanized at 35 weeks of age. All animals were housed under standard conditions (an indoor temperature of 21 °C, relative humidity of 55%, and a 12-h light–dark cycle). All the experiments were conducted at Sun Yat-sen University with the approval of its Institutional Animal Care and Use Committee (SYSU-IACUC-2018-000269).
For the 1,25(OH)2D administration model, the mice were switched from a regular diet to an HFD starting at age 5 weeks. At 16 weeks of age, the mice were randomly divided into two groups: the HFD + 1,25(OH)2D administration group (1,25VD) and the HFD + blank administration group (Veh), with five mice in each group. 1,25(OH)2D powder (AbMole) was dissolved in corn oil (Aladdin) to a concentration of 0.5 µg/mL. The mice in the 1,25VD group were intraperitoneally injected with the 1,25(OH)2D solution every 3 days at a dose of 0.1 µg/kg per injection. The mice in the Veh group received intraperitoneal injections of the same volume of corn oil (vehicle) every 3 days. Tissue samples were collected in the 19th week after administration. All animals were housed under standard conditions (an indoor temperature of 21 °C, a relative humidity of 55%, and a 12-h light–dark cycle). All the experiments were conducted at Sun Yat-sen University with the approval of the Institutional Animal Care and Use Committee (SYSU-IACUC-2021-000637).
Measurement of serum TGs
After the mice were euthanized, whole-blood samples were collected via the tail vein into regular centrifuge tubes. The tubes were then placed at 4 °C and allowed to clot for 2 h. Afterward, the samples were centrifuged at 2 000 × g for 5 min to obtain the serum supernatant. A liquid sample TG assay kit (Applygen Technologies, Inc.) was used according to the manufacturer’s instructions. The samples were incubated at 37 °C for 15 min in a water bath. A microplate reader (BioTek) was used to measure the absorbance at a wavelength of 550 nm. A standard curve was generated based on the OD values obtained from the blank tube and the standard samples, allowing for the calculation of TG levels in the serum samples.
µCT
The mandible and femur samples were fixed using 4% paraformaldehyde (Wuhan Servicebio Technology) for 24 h. Subsequently, µCT analysis was performed using a µCT scanner (SCANCO Medical). Three-dimensional reconstructions of the mandible and femoral bone were generated using Mimics Research 20.0 software. µCT 50 SCANCO MEDICAL software was used to analyze and measure the bone parameters.
Histology staining
Mandible and femur samples were fixed in 4% paraformaldehyde for 24 h. After rinsing with PBS 2-3 times, the samples were decalcified with Tri-EDTA decalcification solution (Wuhan Servicebio Technology) for 3 weeks. The samples were embedded in paraffin for the following section. Paraffin sections (4 μm thick) were subjected to TRAP staining (Sigma), immunofluorescence staining, or Goldner’s trichrome staining (Wuhan Servicebio Technology) according to the provided instructions. TRAP-stained sections and Goldner’s trichrome-stained sections were observed under a light microscope (Leica). For the Goldner’s trichrome-stained sections, unmineralized osteoid stained red, and mineralized bone stained green. The osteoid area and mineralized area of the cortical bones in the femurs were quantified using ImageJ software (National Institutes of Health). The ratio of the osteoid area to the total bone area was calculated for three randomly chosen areas throughout each bone section for each sample, as described by Yuan et al.87
For immunofluorescence staining, paraffin sections were deparaffinized, antigen repaired with sodium citrate, permeabilized in PBST (1x PBS, 0.2% Triton X-100) for 10 min at room temperature and incubated in sheep serum for blocking. Primary antibodies were incubated with the sections overnight at 4 °C, followed by incubation with secondary antibodies overnight at 4 °C. Nuclei were counterstained with DAPI. Images were acquired using a confocal microscope (Olympus). The primary antibody used in this study was an anti-NCAD antibody (Zen-Bioscience) at a 1:50 dilution. Alexa Fluor-coupled secondary antibodies were used (Cell Signaling Technology) at a dilution of 1:500.
BMSC isolation and culture
BMSCs were isolated from the HFD, ND, Veh, and 1,25VD groups as described by Tencerova et al.88 with slight modifications. After the mice were euthanized, the limbs were dissected, after which the bones were removed and then washed with PBS. Using ophthalmic scissors, the ends of the bone epiphyses were trimmed, and the bone marrow tissue was flushed from the bone marrow cavity. The bones without bone marrow were cut into bone fragments measuring 1–3 mm2, which were subsequently digested with a digestion solution containing 1 mg/mL type II collagenase (Gibco) prepared in DF12 medium (Gibco) in a 37 °C shaking incubator at 180 r/min for 5 h. After digestion, the bone pieces were transferred to sterile culture dishes and cultured in a complete medium consisting of DF12 medium and 10% FBS (Gibco). The medium was changed every three days until the cells reached 80% confluence. The adherent cells were dissociated into single cells using TrypLE (Gibco). After centrifugation at 1 000 r/min for 5 min, the supernatant was removed, and 100 μL of PBS was added to gently resuspend the cell pellet, forming a single-cell suspension. The suspension was then mixed with antibodies against CD45-APC (BioLegend) and CD34-APC (BioLegend) and incubated on ice in the dark for 30 min. The reaction was terminated by adding PBS with 10% FBS, and the samples were centrifuged at 4 °C and 1 000 r/min for 5 min. Next, 500 μL of PBS was added to resuspend the cells, and negative selection was performed using a flow cytometer (Becton, Dickinson and Company). The sorted BMSCs were seeded in suitable culture dishes and cultured in a complete medium. The medium was changed every three days until the cells reached 80% confluence, after which the cells were passaged. FACS analysis revealed that these cells were homogeneously positive for the mesenchymal markers CD90 (CD90-PE Ab, BioLegend), CD44 (CD44-PE Ab, BioLegend) and CD29 (CD29-APC Ab, BioLegend) and the stem cell marker Sca-1 (CD34-BV421 Ab, BioLegend) but negative for the hematopoietic markers CD34 (CD34-APC Ab, BioLegend) and CD45 (CD45-APC Ab, BioLegend). The morphology of the BMSCs, which adopted a fusiform, homogenous fibroblastic morphology, was observed under a light microscope. Further differentiation assays showed that these cells could readily differentiate into osteoblasts and adipocytes (Fig. S7). These cells expressed MSC markers and multilineage differentiation potential, aligning with the definition of BMSCs.89 The BMSCs used in this study were obtained from the third passage.
For the induction of senescence models and in vitro transfection experiments, mouse BMSCs were purchased from Cyagen Biosciences and maintained in DF12 medium supplemented with 10% FBS at 37 °C and 5% CO2. A 1,25(OH)2D solution was prepared by dissolving the powder in anhydrous ethanol (Aladdin). After the cells were seeded in a new plate and incubated for 24 h, the following substances were added to the respective groups under light-protected conditions: alcohol + 10% BSA (Veh group), 100 nmol/L 1,25(OH)2D + 10% BSA (1,25VD group), alcohol + 50 μmol/L PA (PA group), 100 nmol/L 1,25(OH)2D + 50 μmol/L PA (PA + 1,25VD group), and 1 mmol/L NAC (AbMole) + 50 μmol/L PA (NAC + 1,25VD group). The cells were incubated under light-protected conditions at 37 °C and 5% CO2 for 24 h. After digestion using TrypLE, the cells were transferred to a new plate for 12 h of subsequent experimental analysis.
Preparation of a PA solution
A PA solution used to induce the senescence model was prepared according to previous methods.90 Briefly, 0.6 g of fatty acid-free BSA (Solarbio) was dissolved in 3 mL of double-distilled water to prepare a 20% fatty acid-free BSA solution. A 0.2% NaOH solution was prepared by dissolving 0.02 g of NaOH in 10 mL of ultrapure water. Then, 0.015 36 g of PA (Sigma) was added to 3 mL of the NaOH solution, and the mixture was saponified in a water bath at 75 °C for 30 min to obtain a 20 mmol/L palmitate saponification solution, which was mixed with the 20% fatty acid-free BSA solution at a 1:1 ratio to obtain a 10 mmol/L PA solution for subsequent experiments. A 10% BSA solution was prepared by mixing the 20% fatty acid-free BSA solution with an equal volume of the 0.2% NaOH solution.
RNA isolation and real‐time RT‒qPCR
Total RNA was extracted from cultured MSCs using NucleoZOL reagent (MACHEREY‑NAGEL) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using the PrimeScript RT Reagent Kit (TaKaRa). Real-time RT‒qPCR was carried out using a Roche Real-time System. Ppia was amplified at the same time to normalize gene expression. Each experiment was repeated three times to determine differences in relative gene expression. The sequences of the PCR primers used in this study are shown in Table S1.
RNA-seq
Following the RNA extraction process described earlier, mRNA was fragmented, labeled, and reverse transcribed into cDNA, which was then purified with the TruSeq RNA Sample Prep Kit (Vazyme) according to the manufacturer’s instructions. The concentration of the resulting cDNA was measured using Qubit fluorometric quantification (Thermo Fisher). Subsequently, the samples were sent to Novogene Biotech Co., Ltd., for sequencing. The raw sequencing data were subjected to quality control analysis, intergroup comparisons, and the generation of gene expression heatmaps and volcano plots using R language.
Next, genes whose expression was significantly upregulated or downregulated in the HFD group compared to that in the ND group at log2FC > 1 and P < 0.05 were chosen for further analyses. Metascape was used to perform GO enrichment analysis of all the differentially expressed genes. Additionally, GSEA software was used to analyze whether specific gene sets, namely, the VDR PATHWAY (C2) and CELLULAR SENESCENCE (C5) gene sets, exhibited differential expression between the groups.91,92
ATAC-seq
The viability of the digested cells was evaluated using trypan blue (Gibco), with the viability threshold set at 85% or higher. A total of 5 × 104 cells per sample were isolated for ATAC library preparation using the TruePrepTM DNA Library Prep Kit V2 for Illumina® (Vazyme) and the TruePrepTM Index Kit V2 for Illumina® (Vazyme). The cells were washed once with 50 μL of cold PBS and centrifuged at 500 × g for 5 min at 4 °C. The cell pellets were resuspended in 50 µL of ice-cold lysis buffer and incubated on ice for 10 min. The cells were then centrifuged at 4 °C and 500 × g for 5 min to collect the nuclei. Next, 20 µL of a transposase reaction mixture was added, followed by transfer of the suspension to a new PCR tube containing the nuclear pellet. The reaction was carried out at 37 °C for 30 min. The resulting product was purified using DNA purification and magnetic bead purification, and the supernatant was transferred to a new PCR tube. PCR amplification was performed using double-ended adapter primers, PPM, DNA polymerase, and a TAB solution. The product was subjected to quality control through Qubit concentration measurements and fragment distribution analysis using the Bioptic Qsep 100 analysis system. The samples were subsequently sent for sequencing (conducted by Novogene Biotech Co., Ltd.).
After quality control, initial sequence alignment, and the removal of duplicate and mitochondrial sequences, MACS2 was used for peak calling, followed by DESeq2 for intergroup comparisons. Peaks were annotated using ChIPseeker to determine their genomic positions and nearby genes, and volcano plots were generated. The distribution of the ATAC-seq peaks was visualized using deepTools, and all the data were visualized using Integrated Genomic Viewer (IGV) software. Further analysis focused on genes showing differential chromatin accessibility with an absolute log2FC > 1 and P < 0.05. Metascape was used to perform GO enrichment analysis of all the differentially accessible genes. Motif analysis was conducted using HOMER.
Induction of osteogenic and adipocyte differentiation
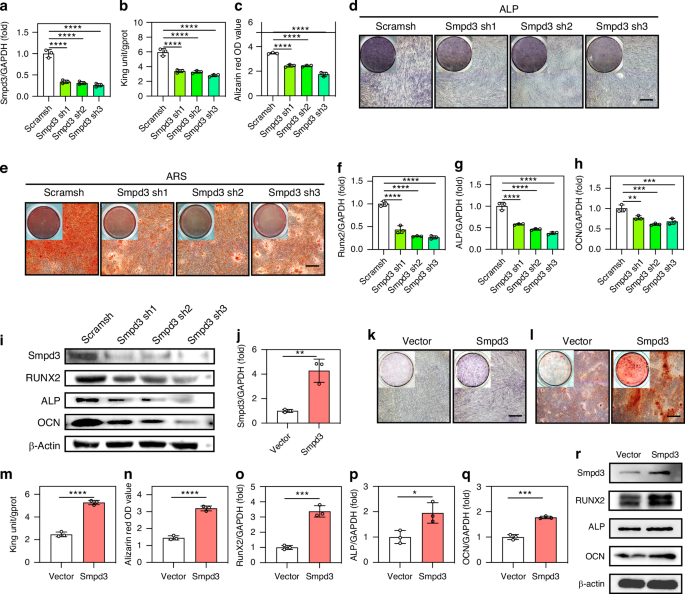
To induce osteogenic differentiation, BMSCs were seeded at a density of 1 × 104 cells per well in a 48-well plate and cultured in DF12 medium supplemented with 10% FBS. When the cell density reached 70%-80%, the medium was replaced with an osteogenic differentiation induction medium. During the first week, the medium was replaced with a fresh mineralization induction medium every 3 days, and during the second week, the medium was replaced every 2 days. After 9 days of osteogenic differentiation induction, RNA was extracted for the detection of osteogenic-related gene expression. After 21 days of osteogenic differentiation induction, the original culture medium was removed, and the cells were washed three times with PBS. Then, 4% paraformaldehyde was added for fixation at room temperature for 15 min, followed by three washes with PBS. The cells were stained with a 1% alizarin red solution (Cyagen Biosciences) for 15 min and then washed three times with ddH2O. Finally, the cells were observed and photographed by light microscopy (Leica). To quantify the ARS staining results, we analyzed the mineralized area relative to the total area by using ImageJ software (National Institutes of Health). An osteogenic differentiation induction medium was purchased from Cyagen Biosciences.
To induce adipocyte differentiation, BMSCs were seeded at a density of 1 × 104 cells per well in a 48-well plate and cultured in a DF12 medium containing 10% FBS. When the cell density reached 70–80%, the medium was replaced with adipocyte differentiation medium (Cyagen Biosciences) according to the manufacturer’s instructions. After 9 days of differentiation, the cells were fixed in 4% paraformaldehyde phosphate buffer for 20 min and washed with phosphate-buffered saline (PBS). An Oil Red O solution (Cyagen Biosciences) was added, and the samples were incubated for 15 min and washed with PBS.
Senescence‐associated‐β‐galactosidase (SA-β-gal) staining
BMSCs were seeded at a density of 1 × 105 cells per well in a 6-well plate and incubated for 24 h, after which the original culture medium was removed, and the cells were washed three times with PBS. Following the instructions of an SA-β-gal assay kit (Beyotime Biotechnology), the cells were fixed at room temperature for 15 min, followed by three washes with PBS. A working solution was prepared and used for staining, and the cells were observed and photographed by light microscopy.
CCK8 assay
BMSCs were seeded at a density of 1 × 103 cells per well in a 96-well plate and cultured in DF12 medium containing 10% FBS. On the 1st, 3rd, 5th, 7th, 9th, and 11th days after seeding, the culture medium was replaced with a mixture of 100 μL of culture medium and 10 μL of CCK8 solution (YEASEN) under light-protected conditions. The cells were incubated at 37 °C and 5% CO2. The absorbance of each well at 450 nm was measured with a microplate reader (BioTek). For each group, triplicate wells were used, and the experiment was repeated at least three times.
EdU staining
After BMSCs were seeded at a density of 1 × 105 cells per well in a 6-well plate and incubated for 24 h, EdU (Beyotime Biotechnology) was added to the culture medium to a final concentration of 10 μM. The cells were then incubated at 37 °C and 5% CO2 for an additional 48 h. Afterward, the original culture medium was removed, and the cells were fixed, permeabilized, treated with reaction solution, and finally stained. The samples were observed and imaged by fluorescence microscopy (Leica).
Immunocytochemical staining
For immunocytochemical staining, cultured cells were fixed in 4% (vol/vol) paraformaldehyde for 30 min at room temperature, permeabilized in PBST (1x PBS, 0.2% Triton X-100) for 10 min at room temperature, and incubated in sheep serum for blocking. Primary antibodies were incubated overnight at 4 °C, followed by incubation with secondary antibodies overnight at 4 °C. The primary antibodies used in this study included anti-p21 (BD Pharmingen) at a dilution of 1:100, anti-γH2A.X (Abcam) at a dilution of 1:500, and anti-VDR (Santa Cruz Biotechnology) at a dilution of 1:50. Alexa Fluor-coupled secondary antibodies were used (Cell Signaling Technology) at a dilution of 1:500.
Western blot
Proteins were extracted from the BMSCs of each group of mice or from in vitro samples. The cytoplasmic and mitochondrial proteins were extracted using the Cytoplasmic and Mitochondrial Protein Extraction kit (Beyotime Biotechnology), according to the manufacturer’s instructions. Immunoblotting was conducted as previously described.93 Primary antibodies against P21 (BD Pharmingen) at 1:500, VDR (Santa Cruz Biotechnology) at 1:500, SOD1 (Santa Cruz Biotechnology) at 1:1 000, SOD2 (Santa Cruz Biotechnology) at 1:1 000, cytochrome C (Zen-Bioscience) at 1:500, AIF (Zen-Bioscience) at 1:500, cleaved caspase-3 (Zen-Bioscience) at 1:500 and Vinculin (Zen-Bioscience) at 1:2 000 were used. Immunoreactive bands were visualized with enhanced chemiluminescence (ECL) (Epizyme Biotech) and analyzed with a ChemiDocTM MP imaging system (Bio-Rad).
Measurement of intracellular ROS levels
Intracellular ROS levels were determined using the oxidation-sensitive fluorescent probe DHE (KeyGEN) according to the manufacturer’s instructions. BMSCs were seeded at a density of 5 × 104 cells per well in a 24-well plate and cultured in DMEM supplemented with 10% FBS for 24 h. Afterward, DHE was added to the medium to a final concentration of 10 µmol/L, and the cells were incubated at 37 °C and 5% CO2 for 30 min. The adherent cells were detached into single cells using TrypLE, followed by centrifugation at 1 000 r/min for 5 min and removal of the supernatant. The cells were resuspended in 200 μL of PBS and analyzed by flow cytometry.
Measurement of SOD activity
BMSCs were seeded at a density of 3 × 105 cells per well in a 12-well plate and cultured in a culture medium for 24 h. Afterward, the cells were scraped using a cell scraper and transferred to a centrifuge tube. Cold PBS was added, and the cells were sonicated on ice to ensure complete cell lysis. Afterward, the mixture was centrifuged at 4 °C and 12 000 × g for 5 min, after which the SOD activity was measured with a Total Superoxide Dismutase Assay Kit (Beyotime Biotechnology). The samples were prepared according to the manufacturer’s instructions and analyzed with a microplate reader.
Luciferase activity assay
Using mouse genomic DNA as a template, the Sod2 promoter region (−200 to +100) was amplified via PCR, which yielded two PCR products. One product contained the predicted VDR-binding site, “CAGGGTCA”, while the other product contained the mutation site, “ACTCCCGC”. These two PCR products were subsequently inserted into the GV238 vector, which contains the firefly luciferase gene, to generate the Sod2-GV238 and Sod2mut-GV238 mutant plasmids, respectively (Fig. S4A). All the abovementioned plasmids were synthesized by GENECHEM.
BMSCs were seeded at a density of 1 × 104 cells per well in a black, opaque 96-well fluorescence plate. After 24 h, Lipofectamine 2000 (Thermo Fisher Scientific) was used to cotransfect the cells with a Renilla luciferase plasmid (internal control), and either the GV238 empty vector (the GV238 vector without the Sod2 promoter region), the Sod2-GV238 plasmid, or the Sod2mut-GV238 mutant plasmid for 12 h. The medium was then replaced with culture medium supplemented with either 10−7 mol/L 1,25(OH)2D or an equal volume of ethanol for another 24 h. The luciferase activity was ultimately analyzed using a dual-luciferase assay kit (Promega Corporation, USA) following the manufacturer’s instructions.
ELISA
BMSCs derived from HFD-fed and ND-fed mice were seeded in six-well plates at a density of 1 × 105 cells per well and incubated for 72 h. The levels of IL6 in the cell supernatants were quantified with commercially available ELISA kits (Cusabio, Wuhan, China) following the manufacturer’s directions.
RNA interference
To suppress endogenous levels of SOD2, BMSCs were transfected with siRNAs targeting SOD2 (RiboBio) using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s protocol. For the analysis of the resulting protein expression levels, the cells were lysed for Western blot analysis at specified time points after transfection.
The cells were initially seeded in antibiotic-free growth medium 24 h before transfection. The transfection solution was prepared as follows: Lipofectamine 2000 at a concentration of 1 mg/mL was diluted to 30 µg/mL in Opti-MEM (31985, Gibco) and mixed for 15 min. Simultaneously, 20 × 10−6 mol/L solutions of each siRNA were diluted to 1 × 10−6 mol/L in a separate aliquot of Opti-MEM. The two solutions were then combined at a 1:1 ratio by volume and incubated for an additional 15 min at room temperature. Subsequently, the final mixture was introduced into the cell culture medium without antibiotics to a final siRNA concentration of 100 × 10−9 mol/L and Lipofectamine 2000 concentration of 3 µg/mL. The cells were incubated with siRNA for 24 h at 37 °C with 5% CO2. Cells treated solely with Lipofectamine 2000 served as the negative control group.
Catalase activity assay
Proteins were extracted from the BMSCs of ND- or HFD-fed mice. The protein concentration was measured using a BCA protein assay kit (CWBIO). The supernatant was subjected to catalase activity analysis with a Catalase Assay Kit (Beyotime) according to the manufacturer’s instructions.
Statistical analysis
All the data were presented as the mean ± SEM of at least three independent experiments. After normality testing, all the data were analyzed by two-tailed unpaired Student’s t-test, the Kruskal–Wallis test (nonparametric sample) or one-way ANOVA (parametric sample) followed by Dunn’s test (nonparametric sample) or Tukey’s post hoc test (parametric sample) as a post hoc test. All the statistical analyses were performed with GraphPad Prism software.









留言 (0)