
記住我








These studies were conducted in accordance with the Declaration of Helsinki and Korean Good Clinical Practice and the International Conference on Harmonization and the study protocol was approved by the Ministry of Food and Drug Safety (MFDS) and the Chungnam National University Hospital Institutional Review Board ([IRB] study 1: IRB No. CNUH 2020-06-031, study 2: IRB No. CNUH 2020-09-117). Both Studies 1 and 2 were registered at ClinicalTrials.gov (study 1: NCT04478097; study 2: NCT04627207). All study participants gave informed consent. The informed consent form was reviewed and approved by the MFDS and the IRB.
SubjectsStudies 1 and 2 recruited 51 and 50 healthy adult volunteers, respectively, aged between 19 and under 55 years, with a minimum weight requirement of 55 kg for men and 45 kg for women. Additionally, the participants were required to possess a body mass index (BMI) within the range of 17.5–30.5 kg/m2. The inclusion of study participants was based on the evaluation of their vital signs, physical examination, clinical laboratory tests, and 12-lead electrocardiography (ECG). Additionally, we collected information on medical history, family history, and current or scheduled medication use through interviews, which was utilized in the screening. The predominant criteria for exclusion included a deviation of blood pressure from the reference range (systolic blood pressure 90–140 mmHg, diastolic blood pressure 60–90 mmHg), measured after 5 min of rest in a seated position, a creatine phosphokinase (CPK) level exceeding three times the upper limit of the reference range, and aspartate transaminase and alanine transferase levels surpassing two times the upper limit of the reference range. Individuals with prior incidences of angioedema as ADRs to ACEIs or ARBs were also excluded. All volunteers provided written informed consent prior to the commencement of the trial.
Study DesignThis study was conducted at the Chungnam National University Hospital Clinical Trial Center (Daejeon, Republic of Korea) in compliance with the principles of the Declaration of Helsinki. The experimental protocols of studies 1 and 2 were identical, except for the variation in the ratios of the drugs administered.
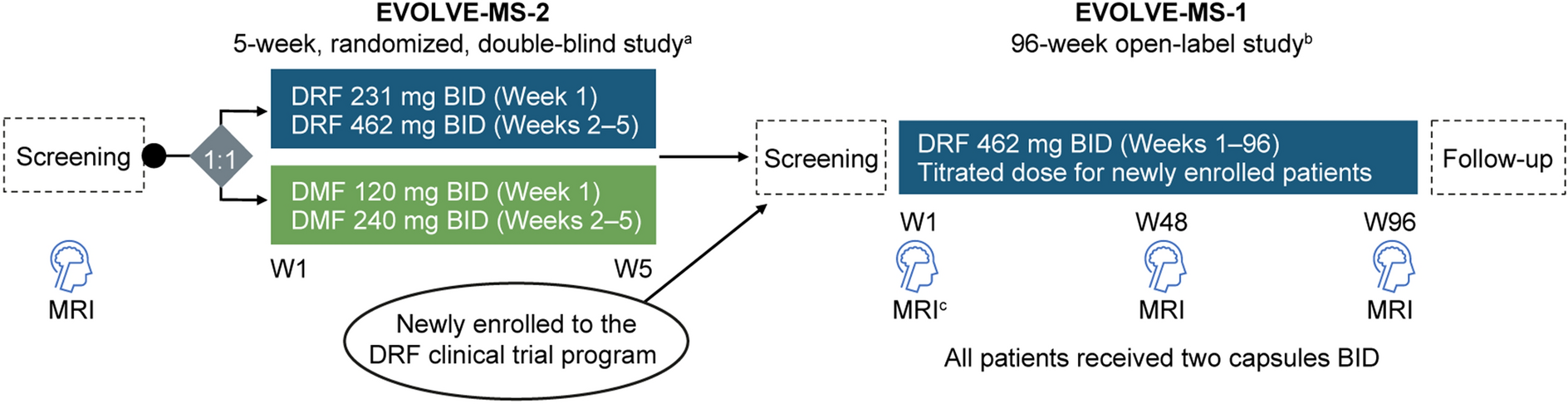
The present investigation constituted a phase 1 clinical trial that employed a randomized, open-label, single-dose, three-sequence, three-period, partially repeated crossover methodology. The study participants were randomized into three sequences in a 1:1:1 ratio. The order of drug administration varied between the distinct sequences; nevertheless, all individuals received two doses of the individual formulations and a single dose of the FDC. The study design is depicted in Fig. 1. The participants fasted for a minimum of 10 h prior to each administration. A 14-day washout period was maintained between each period, the duration of which exceeded the previously reported half-life of the candesartan/amlodipine/atorvastatin by five times [35,36,37].
Fig. 1
Study design. Notes: Individual formulations, individual formulations of candesartan/amlodipine + atorvastatin; fixed-dose combination, fixed-dose combination of candesartan/amlodipine/atorvastatin
Blood was drawn at 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 48, and 72 h post administration and collected in dipotassium ethylenediaminetetraacetic acid anticoagulation tubes. The blood samples were centrifuged at 1950–1980×g for 10 min at 4 °C and the plasma was stored in Eppendorf tubes at temperatures below − 60 °C for subsequent analysis.
Investigational ProductsStudy 1A 16/10 mg Cantabell Tab (16 mg candesartan cilexetil and 10 mg amlodipine; Chong Kun Dang Pharm. Corp, Seoul, Republic of Korea) and a 40 mg Lipitor Tab (40 mg atorvastatin; Pfizer Inc., Seoul, Republic of Korea) were employed as reference drugs (individual formulations). A 16/5/40 mg dose of CKD-333 (16 mg candesartan cilexetil, 10 mg amlodipine, and 40 mg atorvastatin), manufactured by Chong Kun Dang Pharm. Corp (Seoul, Republic of Korea), was used as the test drug (FDC) in study 1. The investigational product, Cantabell Tab, used in these studies was confirmed in previous clinical trials to have pharmacokinetic characteristics bioequivalent to its individual formulations (ClinicalTrials.gov identifier NCT02548286), and no pharmacokinetic interactions have been observed between candesartan cilextil and amlodipine [38]. Therefore, although Cantabell Tab is not an individual formulation in the strict sense, we assumed that it has the same pharmacokinetics properties as the individual formulation.
Study 2A 16/5 mg Cantabell Tab (16 mg candesartan cilexetil and 5 mg amlodipine) and a 20 mg Lipitor Tab (20 mg atorvastatin) were used as reference drugs and a 16/5/20 mg dose of CKD-333 (16 mg candesartan cilexetil, 5 mg amlodipine, and 20 mg atorvastatin) was used as the test drug (FDC) in study 2.
Determination of Plasma ConcentrationsThe bioassays for determining the plasma concentrations of the test and reference drugs were performed at Bioinfra Co., Ltd. (Yongin, Republic of Korea), which is a designated clinical trial sample analysis institution with excellent clinical laboratory practice certification.
CandesartanBlood was drawn for the analysis of candesartan at 0 (pre-dose), 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, and 48 h post dose. The plasma concentrations of candesartan were measured via ultra-high-performance liquid chromatography (UPLC) using a Waters ACQUITY UPLC™ System (Waters, Milford, MA, USA) coupled with a tandem mass spectrometer (Waters Xevo™ TQ MS; Waters, Milford, MA, USA). Samples were prepared by protein precipitation preceding separation and injected into the LC–MS/MS system comprising a 1.7-μm Waters ACQUITY UPLC® BEH C18 column (2.1 mm ID × 50 mm L; Waters, Milford, MA, USA). A multiple reaction monitoring (MRM) mode was used for the quantification of candesartan at m/z 441.00 → 263.18 and candesartan-d5 at m/z 446.10 → 268.20. The linear calibration curve ranged from 5 to 1000 ng/mL. The concentrations of candesartan in the quality control samples were 15, 400, and 750 ng/mL. The determination coefficient (r2) for the calibration curves was ≥ 0.99. The bioassay for candesartan was validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviation was less than 15%.
AmlodipineBlood was drawn for the analysis of amlodipine at 0 (pre-dose), 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, and 72 h post dose. The plasma concentrations of amlodipine were measured using a Waters ACQUITY UPLC™ System coupled with a Waters Xevo™ TQ-XS mass spectrometer. Samples were prepared by protein precipitation prior to separation and injected into the 1.7-μm Waters ACQUITY UPLC® BEH C18 (2.1 mm ID × 50 mm L) column of the LC–MS/MS system. An MRM mode was used for the quantification of amlodipine at m/z 409.20 → 238.15 and amlodipine-d4 at m/z 413.20 → 238.15. The linear calibration curves ranged from 0.05 to 50 ng/mL. The concentrations of amlodipine in the quality control samples were 0.15, 15, and 37.5 ng/mL. The bioassay for amlodipine was validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviation was less than 15%.
Atorvastatin and 2-OH AtorvastatinBlood was drawn for the analyses of atorvastatin and 2-OH atorvastatin levels at 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, and 48 h post dose. The plasma concentrations of amlodipine were measured using a Waters ACQUITY UPLC™ System coupled with a Waters Xevo™ TQ-S mass spectrometer. Samples were prepared by protein precipitation prior to separation and injected into the 1.7-μm Waters ACQUITY UPLC® BEH C18, 1.7 μm (2.1 mm ID × 50 mm L) column of the LC–MS/MS system. An MRM mode was used for the quantification of atorvastatin at m/z 559.26 → 440.26, atorvastatin-d5 at m/z 564.28 → 445.28, 2-OH atorvastatin at m/z 575.26 → 440.26, and 2-OH atorvastatin-d5 at m/z 580.28 → 445.28. The linear calibration curves ranged from 0.1 to 200 ng/mL for atorvastatin and 0.05 to 100 ng/mL for 2-OH atorvastatin. The concentrations of atorvastatin and 2-OH atorvastatin in quality control samples were 0.15, 15, and 37.5 ng/mL, and 0.15, 50, and 75 ng/mL, respectively. The determination coefficient (r2) for the calibration curves was ≥ 0.99. The bioassays for atorvastatin and 2-OH atorvastatin were validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviations were less than 15%.
Pharmacokinetic AnalysisThe pharmacokinetic (PK) analysis set included only those participants who had completed all PK sampling procedures and were not liable for any protocol violations. The PK analyses were performed in compliance with actual blood sampling times. The area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration (AUClast) and the maximum plasma concentrations (Cmax) of candesartan, amlodipine, and atorvastatin were deemed the primary PK endpoints. Conversely, the area under the plasma concentration–time curve from times zero to infinity (AUCinf), time of achievement of maximum plasma concentration (Tmax), the terminal elimination half-lives (t1/2) of candesartan, amlodipine, and atorvastatin, and the AUClast, Cmax, Tmax, and t1/2 for 2-OH atorvastatin were considered the secondary PK endpoints. PK analyses were performed via a non-compartmental method using the WinNonlin® software (version 8.3; Pharsight, Mountain View, CA, USA). AUC was calculated using the linear trapezoidal method. Cmax and Tmax were determined directly from the observed concentration–time data.
Statistical AnalysisStatistical analyses were performed using the SAS® software (version 9.4, SAS Institute Inc., Cary, NC, USA). In the context of the individual formulations group, the PK data recorded in the two distinct periods were regarded as separate datasets, even if they belonged to the same person. All PK parameters were summarized using descriptive statistics. The primary PK endpoints were estimated as the geometric mean ratios (GMRs) and the 90% confidence intervals (CIs) of the FDC to individual formulations. The average bioequivalence (ABE) approach was employed to assess the primary endpoints for candesartan and amlodipine in order to determine whether the 90% CI was constrained within the BE range of 0.8–1.25. Previous reports have portrayed atorvastatin as a potential highly variable drug (HVD), and a mixed-scaled average BE strategy was used in compliance with the local regulatory criteria stipulated by the MFDS (Republic of Korea) [39, 40]. On the basis of the ABE, the confirmation of the 90% CI within the range of 0.8–1.25 was examined to determine the AUClast, independent of the within-subject coefficient of variation of the reference drug (CVwr). To measure Cmax, ABE was employed if the CVwr was less than or equal to 0.3 as calculated from the Cmax of the reference (individual formulations) drugs at two distinct periods. In contrast, if the CVwr was greater than 0.3, the BE range calculated by the reference-scaled average bioequivalence (RSABE) was used to assess whether the 90% CI was limited within the BE range.
SafetyThe safety and tolerability of the test and reference drugs were assessed depending on the incidence of adverse events (AEs), vital signs, physical examination, clinical laboratory tests (complete blood cell count, blood chemistry tests, namely liver and kidney function tests, and urinalysis), and 12-lead ECG of the participants. AEs were coded using the system organ class and preferred term in accordance with MedDRA version 20.1. AEs were segregated into the treatment groups based on the number of individuals, incidence, and number of events. Furthermore, the severity and relationship between AEs were evaluated and summarized.
留言 (0)