
記住我










Retinitis pigmentosa (RP) is a progressive rod-cone dystrophy caused by the dysfunction and eventual death of rod photoreceptors, followed by cone photoreceptor cell death. Initial symptoms of this disease include impaired dark adaptation and “night blindness” (nyctalopia), followed by a loss of peripheral vision and, in advanced cases, possible tunnel vision. As the disease progresses, central vision is also affected (Hartong et al., 2006). RP is currently incurable and is the leading cause of visual impairment and blindness in individuals under the age of 60 (Cross et al., 2022). While clinical therapy cannot reverse the damage caused by RP, efforts are focused on slowing down visual deterioration, treating complications, and helping patients cope with the psychosocial impact of their condition (Frick et al., 2012).
The rhodopsin gene (Rho) was the first gene identified to cause RP. This gene encodes Rhodopsin protein (RHO), a seven-transmembrane G-protein-coupled receptor expressed exclusively in mammalian rod photoreceptors. RHO is synthesized and folded in the endoplasmic reticulum (ER) and processed in the Golgi (Deretic, 2006). Proper folding and trafficking of RHO is essential for normal phototransduction and rod cell survival. The fully functional form of RHO consists of the apoprotein opsin, which binds the chromophore 11-cis retinal (Hargrave, 2001; Pearring et al., 2013). RHO accounts for approximately 90% of all proteins in the discs. Its absence prevents outer segment (OS) formation and leads to photoreceptor cell death (Berson, 1996; Humphries et al., 1997; Lem et al., 1999; Rakshit and Park, 2015).
Rho-mediated RP can be inherited in an autosomal dominant (adRP) or autosomal recessive (arRP) pattern. In the adRP cases, over 150 mutations in the Rho gene have been identified, accounting for 20%–30% of all adRP cases (Athanasiou et al., 2018). Researchers have attempted to categorize these mutations based on their biochemical and cellular properties (Inglehearn et al., 1991; Kaushal and Khorana, 1994; Mendes et al., 2005; Krebs et al., 2010; Rakoczy et al., 2011). Interestingly, most Rho mutations fall into class II, characterized by protein misfolding, ER retention, and OS instability (Athanasiou et al., 2018).
A 3-base pair (bp) deletion in the human Rho gene, deleting one of the two isoleucine (I) at codon 255 or 256 in the sixth transmembrane domain (referred to in this paper as Rho∆I256), has been suggested to be the major cause of adRP in Europe (Inglehearn et al., 1991; Artlich et al., 1992). Rho∆I256 was first discovered in a British family (Inglehearn et al., 1991) and later found in German, Belgian, and Chinese families (Lorenz et al., 1993a; Li et al., 2010). Patients with this mutation show clinical symptoms and signs of RP with an early onset of nyctalopia, a constricted visual field, and even reduced visual acuity or color vision abnormalities (Lorenz et al., 1993a; Li et al., 2010). However, the biochemical changes or associated molecular mechanisms that link the Rho∆I256 mutation to photoreceptor cell death are still not fully understood. According to Sung et al., Rho∆I256 causes biochemical defects associated with class II mutations, with RHO∆I256 protein localizing and accumulating in the ER (Sung et al., 1993). However, the mechanisms underlying how this mutation leads to photoreceptor cell death remain unclear.
It has been reported that cells with dominant Rho mutations undergo cell death as a result of toxic gain-of-function (GOF) and/or dominant-negative (DN) activity (Athanasiou et al., 2018). GOF mechanisms include protein misfolding, aggregation formation, and unfolded protein response (UPR) induction. On the other hand, DN effects of dominant Rho mutations may eliminate one or more functions associated with normal RHO, affecting the maturation, trafficking, and activity of wild-type RHO (RHOWT). RP caused by the DN mechanisms may be due to RHO aggregates that interfere with endogenous RHO maturation (Wilson and Wensel, 2003). These two mechanisms are supported by studies of the RhoP23H (proline to histidine substitution at position 23) class II mutation (Illing et al., 2002; Saliba PMM et al., 2002; Rajan and Kopito, 2005; Griciuc, 2010). RhoP23H is the most common Rho mutation in North America and arguably the most studied one, accounting for 10% of all cases of adRP in the US in patients of Western European origin (Sullivan et al., 2006). The RhoP23H mutant forms ER-bound aggregates and interferes with the UPR system. In addition, these aggregates prevent the proper processing of RHOWT.
According to several studies, Rho∆I256 and RhoP23H exhibit similar phenotypes (Dryja et al., 1990; Lorenz et al., 1993b; Naash et al., 1993; Sung et al., 1993; Kaushal and Khorana, 1994; Daiger et al., 2007), probably because mutations affecting Rho transmembrane or intradiscal domain residues are generally classified as class II mutations (Lorenz et al., 1993b), suggesting that Rho∆I256 may share a similar pathogenetic mechanism with RhoP23H. In previous studies, we could show that RHOP23H aggregate formation is coupled to ER-associated protein degradation (ERAD) (Griciuc et al., 2010a), a major protein processing pathway that is divided into four steps: substrate recognition, ubiquitination, retrotranslocation to the cytosol, and proteasome-mediated degradation (Guerriero and Brodsky, 2012). Further, we found that misfolded RHOP23H is targeted by valosin-containing protein (VCP), a type II AAA ATPase chaperone that processes and earmarks RHOP23H to be transferred from the endoplasmic reticulum (ER) to the proteasome system for degradation (Wang et al., 2004). VCP is the driving force for the retrotranslocation of ER-retained aggregates from the ER to the cytosol for proteasomal degradation (Ye et al., 2001). Studies have shown that VCP interacts with aggregates or ubiquitin-positive inclusions in patients with neurodegenerative diseases (Mizuno et al., 2003; Boeddrich et al., 2006; Gitcho et al., 2009). Our previous research has also shown that inhibition of VCP activity suppresses retinal pathology and preserves retinal structure and function in RhoP23H-associated adRP animal models (Griciuc et al., 2010b; Arango-Gonzalez et al., 2020; Sen et al., 2021a; Sen et al., 2021b). Here, we show that, similar to RhoP23H, degradation of RHO∆I256 aggregates undergoes a VCP-dependent ERAD process. Most importantly, we find that RHO∆I256-containing cytoplasmic aggregates retain RHOWT and interfere with the normal localization or trafficking of RHOWT to the cell membrane, which explains its dominant-negative mechanism. (All abbreviations are shown in Supplementary Table S1).
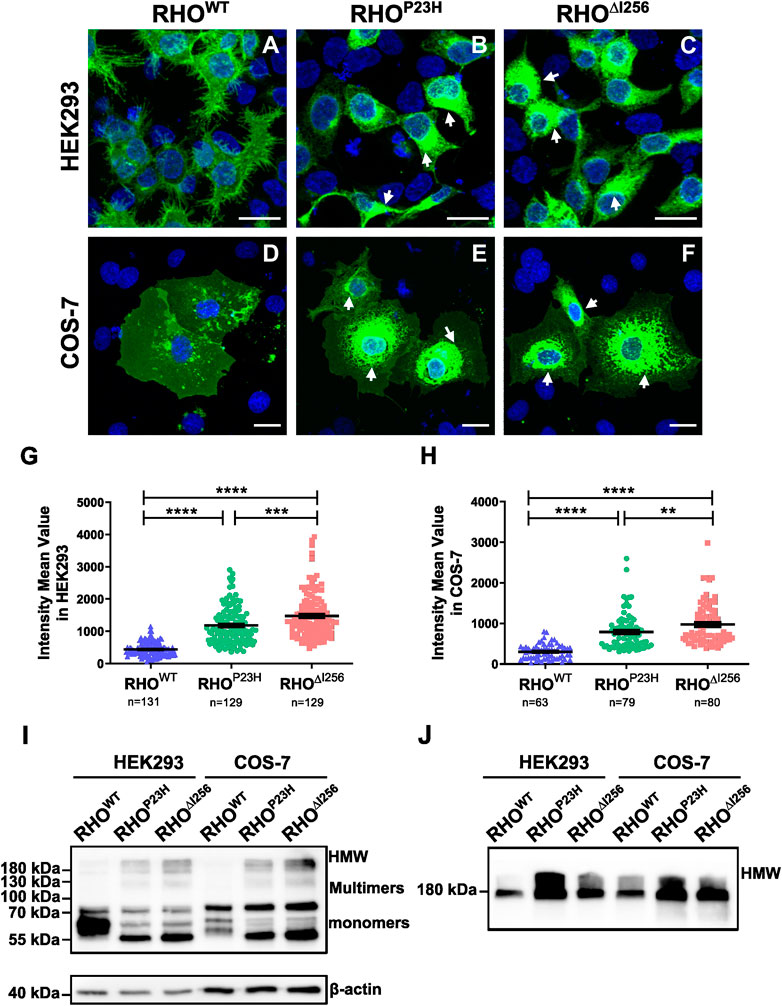
2 Results2.1 Rho∆I256 mutant is mislocalized and tends to form high molecular weight aggregates in the cytosolTo study the cellular behavior of Rho∆I256in vitro, we transfected different Rho constructs (RhoWT, RhoP23H, or Rho∆I256), tagged with EGFP using the pEGFP-N1 vector, into HEK293 and COS-7 cells (Figure 1). As expected, RHOWT-EGFP was correctly targeted and evenly distributed to the plasma membrane in both cell types, with only scattered RHOWT-EGFP puncta in the cytosol, which is consistent with previous results (Figure 1A, D) (Illing et al., 2002; Saliba PMM et al., 2002; Griciuc et al., 2010a). In contrast, most RHO∆I256 was confined to the perinuclear region, with high fluorescence intensity (Figure 1C, F), similar to the cellular localization of RHOP23H (Figure 1B, E). We measured the mean fluorescence intensity (MFI) in the cytoplasmic region for all three groups to quantify RHO aggregation. We found that the MFI of RHO was significantly increased in both Rho∆I256-EGFP and RhoP23H-EGFP transfected cells (HEK293 cells: 1,477 ± 695.9; COS-7 cells: 974.2 ± 503.3; HEK293 cells: 1,182 ± 570.9; COS-7 cells: 788.1 ± 445.8, respectively) compared to RhoWT-EGFP transfected cells (HEK293 cells: 438.8 ± 207.9; COS-7 cells: 303.2 ± 181.6) (Figures 1G, H), indicating that Rho∆I256, similar to RhoP23H, leads to a high degree of RHO accumulation. We also found that Rho∆I256-EGFP results in a significantly higher MFI than RhoP23H-EGFP (HEK293 cells: Rho∆I256 vs. RhoP23Hp < 0.0001; COS-7 cells: Rho∆I256 vs. RhoP23Hp < 0.05, Mann-Whitney-U-test, at least 40 cells in HEK293 and 20 cells in COS-7 cells from three different experiment were quantified), suggesting that the Rho∆I256 mutation induces more severe protein aggregation.
Figure 1. RHO∆I256 mislocalizes to the cytosol and forms high molecular weight aggregates in vitro. Fluorescence images of HEK293 (A–C) and COS-7 cells (D–F) transfected with RhoWT-EGFP, RhoP23H-EGFP, or Rho∆I256-EGFP (green). RHOWT-EGFP is predominantly localized at the plasma membrane, while RHO∆I256-EGFP (white arrows in C, F) and RHOP23H-EGFP (white arrows in B, E) accumulate in the cytosol. Scale bar: 20 μm. There was a significant increase in the mean fluorescence intensity (MFI) in cells transfected with Rho∆I256-EGFP or RhoP23H-EGFP cells compared to RhoWT-EGFP (G–H). Immunoblot analysis revealed that RHO∆I256-EGFP forms HMW aggregates in both soluble (I) and insoluble cell fractions (J). Data were expressed as mean ± SEM. Significance was calculated by Mann-Whitney-U-test. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Given the potential influence of large EGFP tags on the structure, oligomerization, and aggregate formation of the target protein, we transfected HEK293 cells with pEGFP-N1 plasmids encoding only EGFP. We compared the behavior of this control group with that of cells transfected with RhoWT-EGFP, RhoP23H-EGFP, or Rho∆I256-EGFP. Remarkably, cells transfected with the plasmid encoding only EGFP did not exhibit aggregate formation (Supplementary Figure S1), a finding consistent with previous studies (Griciuc et al., 2010a). To further investigate the component of the accumulated RHO aggregates in the cytoplasm, we monitored RHO levels in detergent-soluble (Figure 1I) and detergent-insoluble (Figure 1J) extracts from HEK293 cells that had been transiently transfected with RhoWT-EGFP, Rho∆I256-EGFP, or RhoP23H-EGFP constructs using SDS-PAGE Western blotting. The mature monomeric form of RHO containing the EGFP tag was observed as a band at 55–75 kDa, whereas dimers and trimers appear as smears at approximately 130 kDa and 180 kDa, respectively. High molecular weight (HMW) species with molecular weights greater than 180 kDa indicated RHO-containing oligomers and aggregates. RHOWT-EGFP was mostly detected in soluble extracts (Figure 1I) and showed diffuse bands of the predominantly monomeric form of the mature RHO, with minor multimers or HMW RHO. These results suggest that most RHOWT-EGFP forms a native mature conformation (Griciuc et al., 2010a).
On the other hand, RHO∆I256-EGFP showed a completely different behavior. Most of the mutant RHO was found in dimers, trimers, and HMW oligomers in the soluble cell fraction (Figure 1I), indicating a non-native conformation or immature RHO since RHO is monomeric in its native form in the membrane (Downer and Cone, 1985). Furthermore, a significant amount of RHO∆I256-EGFP appeared as HMW species in the detergent-insoluble fractions (Figure 1J) compared to RHOWT-EGFP, suggesting that Rho∆I256 is more likely to form RHO aggregates than RHOWT. Similar observations were also made for RHOP23H-EGFP.
In conclusion, our results suggest that Rho∆I256, like the RhoP23H mutation, causes a mislocalization of RHO that cannot be effectively targeted to the plasma membrane. In addition, mutant RHO is more prone to form cytosolic non-native HMW aggregates than RHOWT.
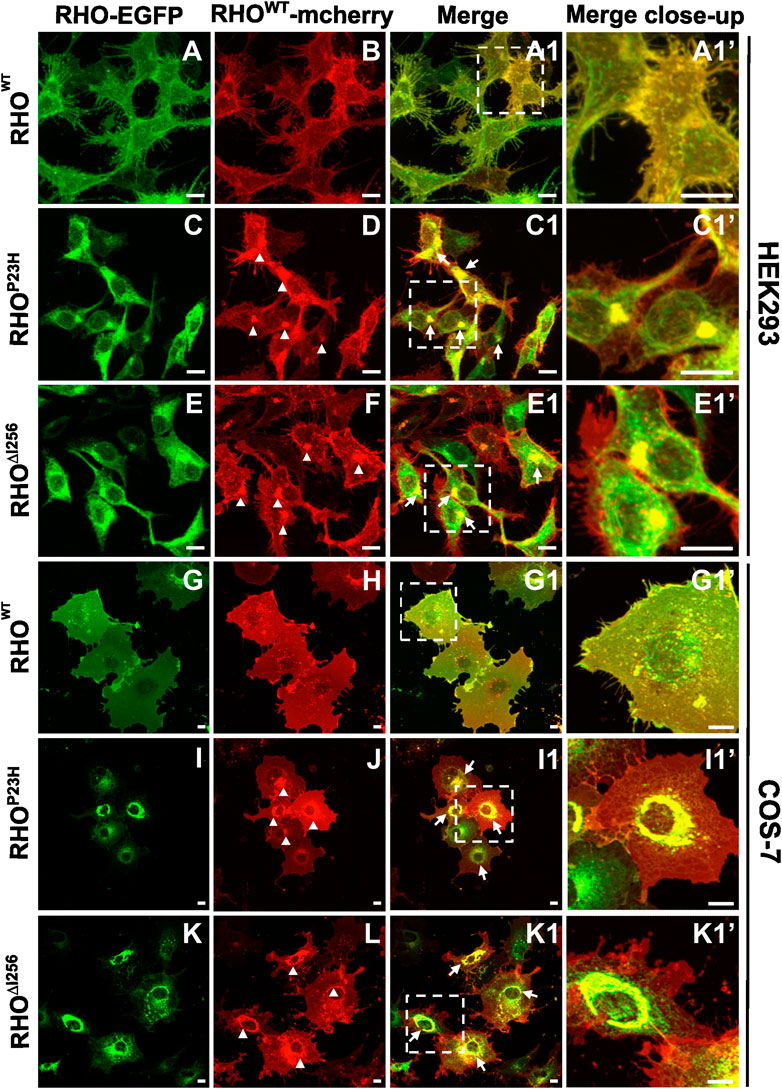
2.2 Dominant Rho∆I256 mutation impairs the localization of WT rhodopsinMost, if not all, patients with adRP carrying the Rho∆I256 mutation inherit one mutant allele and one normal (WT) allele (Inglehearn et al., 1991; Lorenz et al., 1993a; Li et al., 2010). To closely mimic the heterozygous nature of the human patient genotype, we co-transfected HEK293 and COS-7 cells with EGFP-tagged Rho∆I256, RhoP23H, or RhoWT and mcherry-tagged RhoWT at equal concentrations. We used the two fluorescent tags to visualize any colocalization or interaction between mutant RHO∆I256 and RHOWT.
Co-transfection of Rho∆I256-EGFP and RhoWT-mcherry in HEK293 and COS-7 cells resulted in intracellular mislocalization of RHO∆I256-EGFP as previously observed (Figure 2E, K). Here, RHOWT-mcherry was also mislocalized to the perinuclear region and overlapped significantly with Rho∆I256-EGFP aggregates (Figure 2E1, K1). In comparison, when co-expressing both differently tagged RhoWT plasmids, RHOWT-EGFP and RHOWT-mcherry were predominantly localized to the plasma membrane (Figure 2A, B, G, H), as previously observed for RHOWT. These results indicate that RHO∆I256 impairs the correct membrane localization of the RHOWT protein. Since a fraction of RHOWT-mcherry was still targeted to the cell membrane in cells co-transfected with mutant RHO, this suggests that the impairment of RHOWT was not complete. Similar results were obtained in cells co-expressing RHOWT-mcherry with RHOP23H-EGFP (Figures 2I,J), indicating that the molecular pathomechanisms underlying the two dominant Rho mutations are likely similar.
Figure 2. Rho∆I256-EGFP and RhoWT-mcherry co-expression causes intracellular retention of WT rhodopsin. Fluorescence microscopy detecting EGFP (A, C, E, G, I, K) and mcherry (B, D, F, H, J, L) in HEK293 and COS-7 cells co-transfected with RhoWT-mcherry and Rho∆I256-EGFP or RhoP23H-EGFP plasmids. (A1, C1, E1, G1, I1, and K1) show the merged images of the EGFP and mcherry channels. Arrows indicate the colocalization of RHOWT-mcherry with RHO∆I256-EGFP or RHOP23H-EGFP. Magnified view is shown in (A1', C1', E1', G1', I1', and K1'). Arrowheads mark the mislocalized RHOWT retained in misfolded aggregates (D, F, J, L). Scale bar: 10 μm.
Our results show that mislocalized RHO∆I256 aggregates trap part of RHOWT in the aggregate pool, preventing RHOWT from reaching its correct location in the cell membrane. As a result, a dominant-negative (DN) effect may play an important role in the pathology of Rho∆I256-associated retinal degeneration (Herskowitz, 1987).
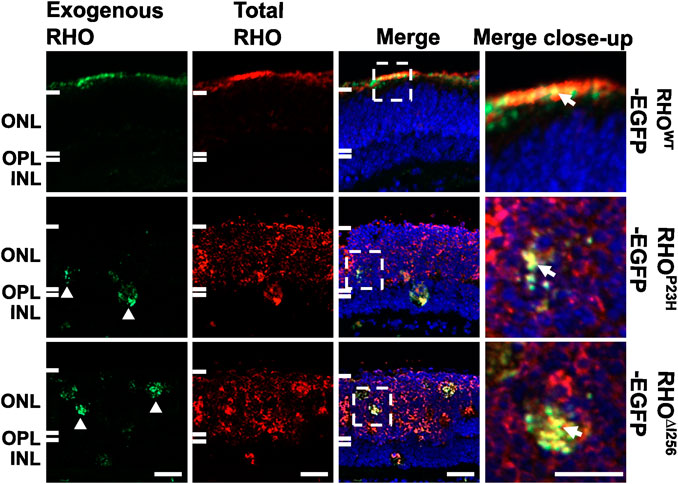
2.3 Proper targeting of endogenous WT rhodopsin to the photoreceptor outer segment is compromised by exogenously expressed RHO∆I256 in C57BL/6J retinal explantsThe recruitment of RHOWT into the RHO∆I256 aggregates in cells supports the hypothesis that the Rho∆I256 allele causes RP via a DN mechanism. To investigate this, we transfected Rho∆I256-EGFP into retinal explants from C57BL/6J (wild-type, WT) mice, which naturally produce RHOWT that localizes to the rod outer segments (ROS). To determine whether RHO∆I256 affects endogenous RHOWT localization, we used reverse magnetofection, a technique to deliver Rho∆I256-EGFP into the C57BL/6J retina (Gomez-Tourino et al., 2013; Sen et al., 2021a; Bassetto et al., 2021). We labeled total RHO with a specific red fluorescing dye conjugated antibody to differentiate between EGFP-tagged exogenous RHO (displayed fluorescence in both red and green) and endogenous RHO (exhibited only red fluorescence). The comprehensive details on the Rho∆I256-EGFP transfection group as well as other transfection groups, are listed in Supplementary Table S2.
When C57BL/6J retinal explants were transfected with the RhoWT-EGFP/XPMag complex, both endogenous and exogenous RHOWT correctly targeted the OS layer and largely overlapped each other. However, when the C57BL/6J retinal explants were transfected with the Rho∆I256-EGFP/XPMag complex, a significant amount of endogenous RHOWT was mislocalized and remained in the ONL instead of being properly targeted to the OS (Figure 3). As expected, exogenous RHO∆I256-EGFP was trapped in the ONL and overlapped with part of endogenous RHOWT. We observed the same result in the RhoP23H-EGFP/XPMag-transfected C57BL/6J retinal explants. Furthermore, transfection of either of these two dominant Rho mutants into C57BL/6J retinal explants resulted in reduced OS length (Figure 3), which is a hallmark of retinal degeneration, indicating a degenerative effect (Milam et al., 1996; Sakai et al., 2003). Endogenous RHO was correctly targeted to the OS in all three control groups (complete medium CM, XPMag, or EGFP-transfected groups, as shown in Supplementary Figure S2).
Figure 3. In C57BL/6J retinal explants, the trafficking of endogenous RHOWT is affected by transfection with Rho∆I256. Immunofluorescence pictures reveal the localization of total RHO (red, second column), including endogenously expressed RHO and EGFP-tagged exogenously expressed RHO in three experimental groups in C57BL/6J retinae transfected with RhoWT-EGFP/XPMag (first row), RhoP23H-EGFP/XPMag (second row), and Rho∆I256-EGFP/XPMag (third row). Exogenously expressed RHO (green, first column) is indicated by EGFP fluorescence. Exogenous RHO∆I256-EGFP is mislocalized in the ONL (white arrowheads, green channel) and leads to the mislocalization of endogenous RHOWT (red, third row). Moreover, in Rho∆I256-EGFP/XPMag transfected C57BL/6J mouse retinal explants, mislocalized RHO∆I256-EGFP partially overlaps endogenous RHOWT (white arrows, merged close-up), and the OS layer is thinner. In contrast, exogenous RHOWT-EGFP correctly targets the OS layer and colocalizes with endogenously expressed RHOWT in transfected C57BL/6J mouse retinal explants. Scale bar: 20 μm.
Our findings support the hypothesis that the transport of endogenous RHOWT is impaired in the presence of the dominant Rho∆I256 (or RhoP23H) mutant, suggesting that the DN effect plays an essential role in the pathophysiology of Rho∆I256-associated adRP.
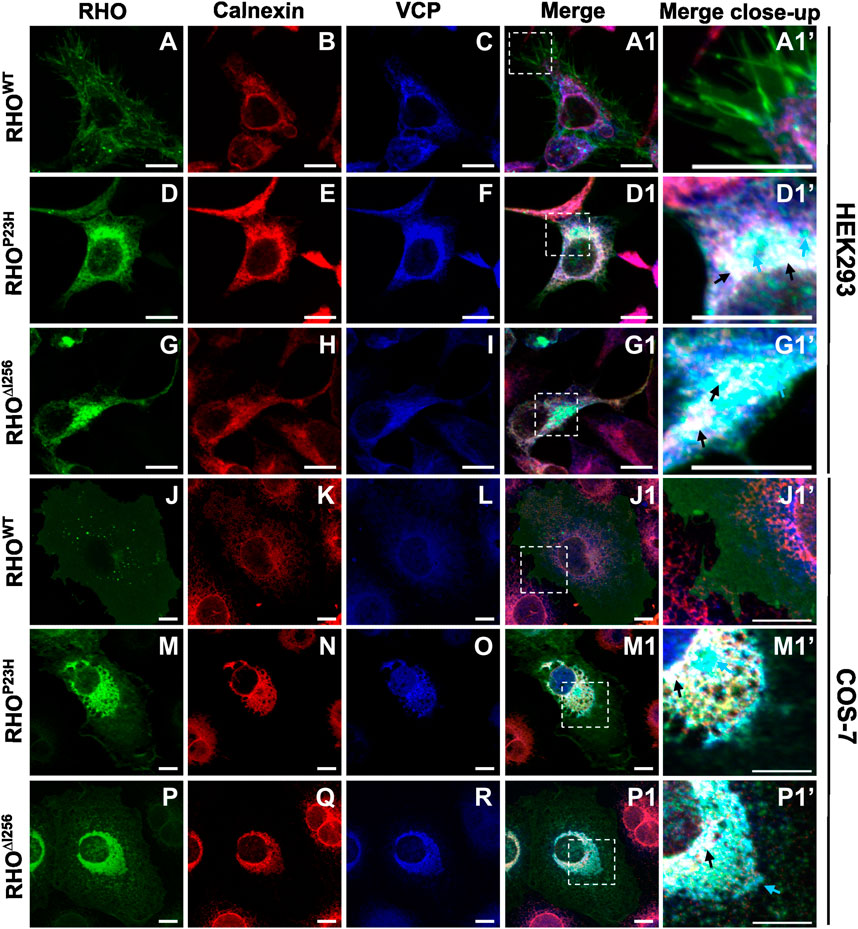
2.4 RHO∆I256 is retained at the ER and forms a complex with the ERAD effector VCPOur results show that the dominant Rho∆I256 mutation leads to cytoplasmic aggregates retaining WT rhodopsin from reaching its final physiological destination at the membrane. We and others have previously shown that such aggregates can be substrate to ERAD, the major degradation pathway for unfolded or misfolded protein aggregates. To determine whether RHO∆I256 aggregates are substrates of the ERAD clearance pathway, we analyzed whether they are tethered to ER sites and recognized by the ERAD effector VCP (Griciuc et al., 2010a; Sen et al., 2021b).
We transfected HEK293 or COS-7 cells with plasmids encoding EGFP-tagged Rho∆I256, RhoP23H, or RhoWT and analyzed colocalization of RHO-EGFP with ER or VCP by immunofluorescence staining with calnexin- or VCP-specific antibodies. Our results revealed a partial colocalization between RHO∆I256 aggregates with ER and VCP in both cell lines (Figure 4). In RhoWT-EGFP transfected cells, we hardly observed any colocalization of RHOWT with calnexin or VCP (Figure 4A1’, J1’). In contrast, in Rho∆I256-EGFP transfected cells, aggregated mutant RHO∆I256 partially colocalized with the ER marker calnexin and with VCP (Figure 4G1, P1 and black arrows in G1’, P1’). Furthermore, some of the RHO∆I256-containing cytoplasmic aggregates were not found within the ER but only co-stained with VCP, indicating that they were extracted from the ER and delivered to the cytosol by VCP. Similarly, the RhoP23H mutation led to the formation of aggregates that colocalized with calnexin and VCP, in agreement with our previous results (Griciuc et al., 2010a). Using Pearson’s correlation coefficient analysis, we further quantified the colocalization between RHO and ER or VCP (Supplementary Figure S3A, B). Compared with RHOWT protein, RHO∆I256 and RHOP23H aggregates showed significantly increased association with ER or VCP, which was consistent with the above immunostaining data.
Figure 4. RHO∆I256 aggregates colocalize with ER and VCP. Immunofluorescence staining shows the localization of transfected Rho-EGFP (green, first column), the ER marker Calnexin (red, second column), and endogenous VCP (blue, third column) in HEK293 (A–I, A1, D1, G1) or COS-7 cells (J–R, J1, M1, P1). Colocalization is revealed in merged pictures (fourth column), and higher magnification pictures from merged insets are shown in column 5. RHOWT is mainly localized to the plasma membrane (A and J). RHO∆I256 aggregates localize with VCP to the ER (black arrows, D1’, M1’) and partially colocalize only with VCP (cyan arrows, D1’, M1’). RHOP23H also accumulates in ER and VCP (black arrows, G1’, P1’). Scale bar: 10 μm.
The localization of endogenous VCP was observed predominantly in the cytoplasm in RhoWT-EGFP-transfected cells (Figure 4C, L). However, in cells that express mutant RHO∆I256 (Figure 4I, R) or RHOP23H (Figure 4F, O), the distribution of VCP mostly colocalized with calnexin, suggesting that the recruitment of VCP to the ER is involved in the retrotranslocation of aggregates.
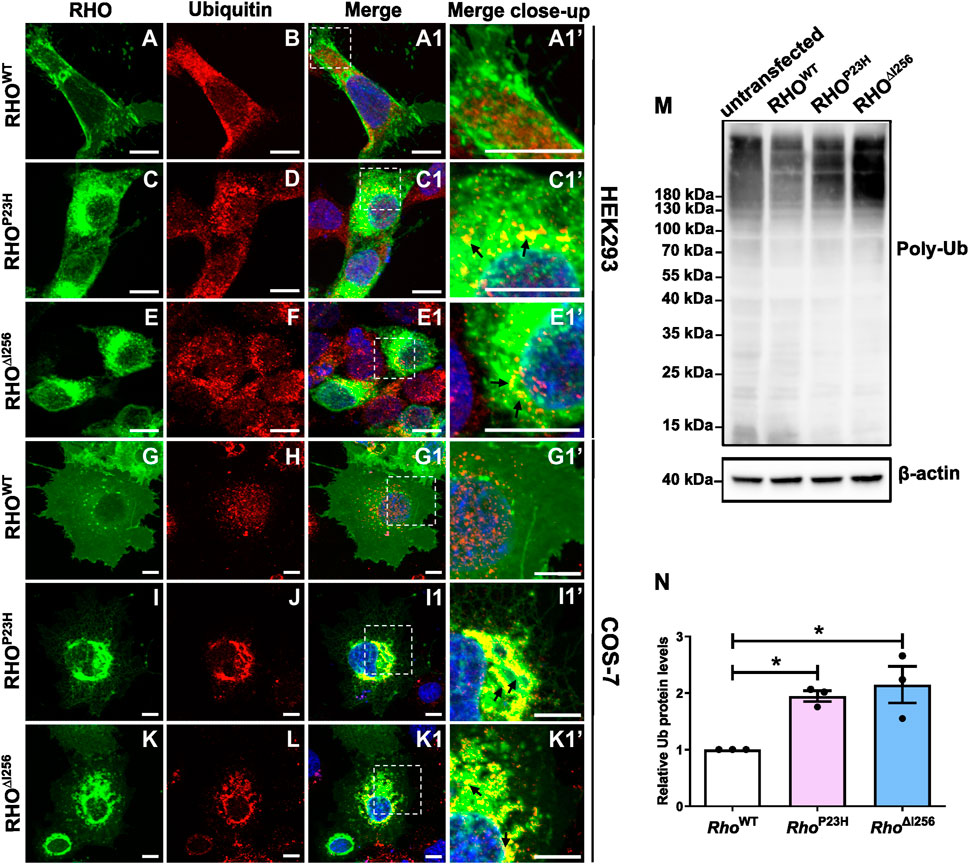
2.5 Mislocalized RHO∆I256 aggregates are partially polyubiquitinatedIt is known that almost all ERAD substrates are ubiquitinated prior to targeting the proteasome (Illing et al., 2002). In order to evaluate the role of ubiquitin (Ub) in the degradation of RHO∆I256 aggregates or RHOWT, we compared the changes in the localization and expression levels of ubiquitin proteins in Rho∆I256-EGFP or RhoWT-EGFP transfected cells using ubiquitin-specific antibodies. RhoP23H-EGFP transfected cells were used as a positive control since RHOP23H is much more ubiquitinated than RHOWT (Illing et al., 2002).
We found that RHO∆I256 aggregates were strongly ubiquitinated in transfected HEK293 and COS-7 cells colocalizing with endogenous ubiquitin (Figure 5). In both cell types transfected with RhoWT-EGFP, ubiquitin was not specifically colocalized with rhodopsin, yet distributed throughout the whole cytosol (Figure 5B, H). No enhanced colocalization between RHOWT and ubiquitin was observed in merged images (Figure 5A1, G1). In contrast, in Rho∆I256-EGFP transfected HEK293 cells, ubiquitin staining was found in numerous accumulated puncta (Figure 5F), which localized with RHO aggregates in the cytosol (Figure 5E1’, C1’). In COS-7 cells, the ubiquitin staining pattern was less punctated, more clustered (Figure 5L), and was mainly recruited to the RHO aggregates pool (Figure 5K1’, I1’). A similar ubiquitin pattern to that in Rho∆I256-EGFP transfected was seen in RhoP23H-EGFP transfected cells. Consistent with these immunostaining results, Pearson’s correlation coefficient analysis also revealed a significantly higher colocalization between RHO∆I256 (or RHOP23H) aggregates and Ub in contrast to RHOWT (Supplementary Figure S3C).
Figure 5. Misfolded RHO∆I256 is ubiquitinated. (A–L) Immunofluorescence of EGFP-tagged RHOWT, RHO∆I256, or RHOP23H (green, first column) and Ub (red, second column) in HEK293 (A–F, A1, A1’-E1, E1’) and COS-7 cells (G–L, G1, G1’-K1, K1’). Colocalization is shown in merged pictures (third column). Ub is recruited into the aggregates pool, which is revealed as yellow dots in merged pictures in Rho∆I256 (black arrows, E1’, K1’) or RhoP23H transfected cells (black arrows, C1’, I1’). Scale bar: 10 μm; (M). Western blot results show enhanced smear Ub bands in cell lysates of Rho∆I256 or RhoP23H compared to RhoWT-transfected HEK293 cells. Ub expression is also present in untransfected control group. β-actin is used as housekeeping marker. (N). Quantification of Ub expression. Data are expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by Bonferroni’s multiple comparison test (*p < 0.05). Three independent experiments were included in the data analysis.
Subsequently, we used immunoblotting to examine the expression pattern of ubiquitin in lysates of untransfected HEK293 cells and in cells transfected with different Rho constructs (untransfected cells were used as control) and demonstrated that similar to RHOP23H, RHO∆I256 is ubiquitinated to a greater extent than RHOWT. We detected a significantly increased intensity of the characteristic ubiquitin smear bands in lysates obtained from mutant Rho∆I256 or RhoP23H transfected cells compared to those obtained from RhoWT transfected cells (Figure 5M), particularly evident in polyubiquitin chains larger than 180 kDa. The untransfected control cells also showed the same pattern of Ub expression, indicating that the ubiquitin signal is not due to overexpression. To quantify the expression of polyubiquitin protein, we compared the levels between WT transfected group and the mutant transfected groups (protein level higher than 20 kDa). The results showed that polyubiquitin was significantly increased in HEK293 cells expressing RHO∆I256 and RHOP23H than in RHOWT (Figure 5N). As ubiquitin acts as a tag that identifies proteins for ERAD-mediated degradation, our results indicate that ubiquitinated RHO∆I256 aggregates are likely to be a substrate of the ERAD pathway.
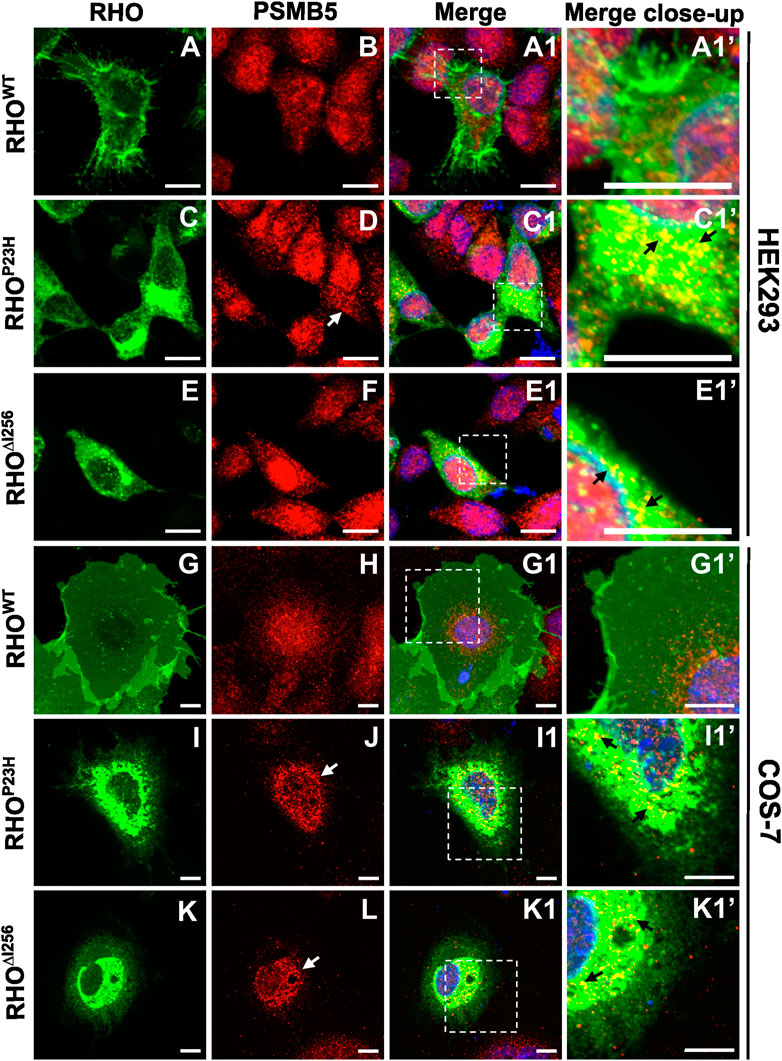
2.6 RHO∆I256 colocalizes with the proteasome 20S subunitThe process of ERAD culminates with the degradation of misfolded proteins by the ubiquitin-proteasome system (UPS). In this process, substrates are tagged with ubiquitin (Ub), form polyubiquitinated adducts, and are then recognized and degraded by the 26S proteasome (Bence et al., 2001; Adams, 2003; Schwartz and Ciechanover, 2009; Sen et al., 2021b). The 26S proteasome consists of one 20S protein subunit and two 19S regulatory cap subunits. The 20S core region is responsible for substrate degradation (Gomez-Tourino et al., 2013). To analyze proteasomal rhodopsin degradation, we used a specific antibody against the proteasome 20S subunit beta 5 (PSMB5) to visualize this subunit in HEK293 and COS-7 cells transfected with plasmids encoding Rho and to observe its colocalization with RHO∆I256 aggregates.
We found that RHO∆I256 aggregates were transported to the proteasome system and colocalized with PSMB5. In RhoWT-EGFP-transfected HEK293 and COS-7 cells, PSMB5 staining was shown in the cytoplasm and the nucleus (Figure 6B, H). However, in RHO∆I256-EGFP-expressing cells, PSMB5 accumulated and formed perinuclear punctate aggregates in the cytosol. This phenomenon was more pronounced in COS-7 cells than in HEK293 cells (Figure 6D, F, J, L). Most importantly, the punctate distribution of PSMB5 overlapped with a fraction of perinuclear RHO∆I256 aggregates (Figure 6C1’, I1’, J1’, K1’). Pearson’s correlation coefficient analysis also showed a significantly greater colocalization between RHO∆I256 (or RHOP23H) aggregates and PSMB5 than RHOWT (Supplementary Figure S3D). These results suggest that a fraction of these aggregates have already been retrotranslocated from the ER to the proteasome system, probably as a consequence of ERAD-mediated degradation.
Figure 6. Rho∆I256 is targeted to the proteasome system. Co-staining of EGFP-tagged RHO∆I256, RHOWT, or RHOP23H (green, first column) with PSMB5 (red, second column) in HEK293 (A–E1’) and COS-7 cells (G–K1’). Colocalization is shown in merged pictures (third column). PSMB5 distributed in the cytosol and nucleus in RhoWT-EGFP transfected cells with no overlapping signal. In Rho∆I256-EGFP transfected cells, PSMB5 partially accumulated in the cytosol (white arrows in F and L) and colocalized with RHO∆I256 aggregates (black arrows in E1’ and K1’). Similar results were also seen in RhoP23H-EGFP transfected cells (white arrows in D and J; black arrows in C1’, I1’). Scale bar: 10 μm.
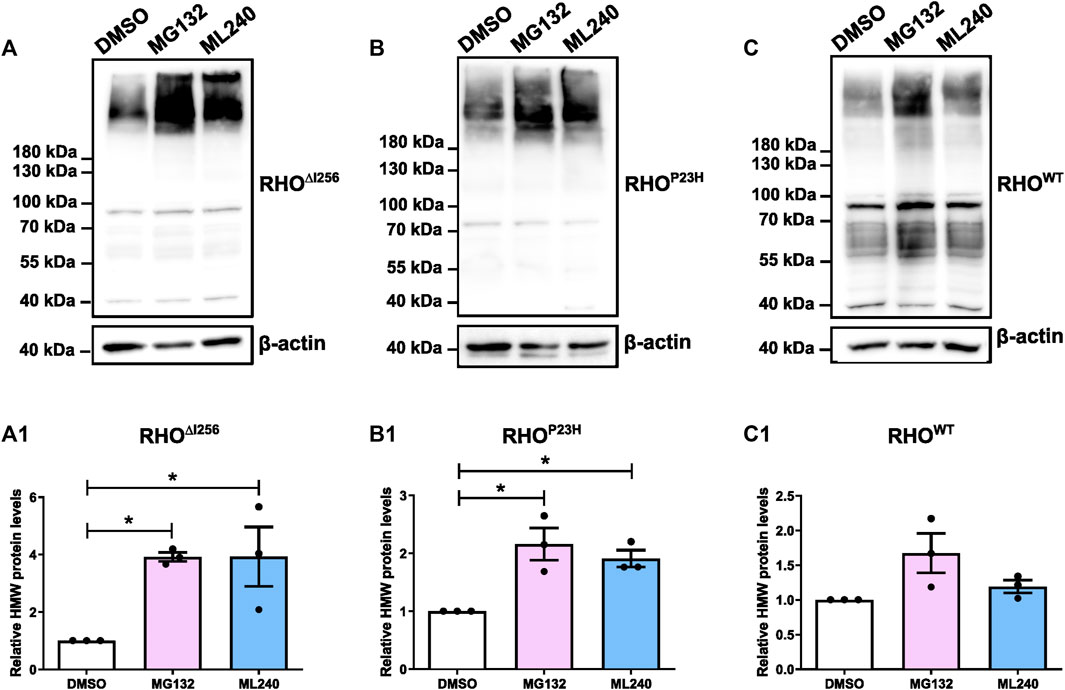
2.7 Inhibition of VCP- and proteasomal activity increase the abundance of high molecular weight RHO aggregates in vitroThe colocalization of RHO∆I256 with different ERAD pathway markers suggested that the ERAD machinery may likely be involved in the clearance of RHO∆I256 aggregates. To verify this hypothesis further, we inhibited the proteasome or the chaperone VCP.
HEK293 cells overexpressing RhoWT-EGFP, Rho∆I256-EGFP, or RhoP23H-EGFP were treated with the proteasome inhibitor MG132 (5 µM), the VCP inhibitor ML240 (2 µM) or vehicle (DMSO, 0.1%), for 6 h. Inhibition with MG132 or ML240 significantly increased the levels of HMW aggregates in the lysates of Rho∆I256-EGFP transfected cells (Figure 7A). The average intensity of the bands corresponding to RHO HMW aggregates (molecular weight≥180 kDa), indicated that MG132 or ML240 significantly increased the amount of HMW proteins compared to the vehicle-treated group. These results suggest that both proteasomal and VCP activity are essential for the degradation of RHO∆I256 during the ERAD process (Figure 7A1).
Figure 7. Proteasome and VCP inhibition increases HMW aggregates load in Rho∆I256-transfected HEK293 cells. Western blot results for RHO in HEK293 cells after transfection with Rho∆I256-EGFP (A), RhoP23H-EGFP (B), or RhoWT-EGFP (C) followed by treatment with DMSO, 5 µM MG132 or 2 µM ML240 for 6 h. The gray value changes in HMW rhodopsin aggregates bands above 180 kDa following ML132 or ML240 treatment relative to DMSO vehicle treatment is quantified and presented in (A1, B1, and C1). β-actin serves as housekeeping protein control. Data were expressed as mean ± SEM. Significance was calculated by a one-way ANOVA and Bonferroni’s multiple comparisons test. *p < 0.05. Three independent experiments are included in this data analysis.
The levels of RHOP23H HMW aggregates also significantly increased after MG132 or ML240 treatment, which is consistent with previous findings (Figure 7B, B1) (Illing et al., 2002; Griciuc et al., 2010a; Griciuc et al., 2010b). In addition, the RHOWT HMW aggregates increased slightly, suggesting that RHOWT, when overexpressed, also requires the proteasome for degradation (Figure 7C, C1).
3 DiscussionRho mutations, a principal cause of adRP, result in changes in protein structure and function. They are classified into seven classes, each with varying defects, including misfolding and disruptions in proteostasis, mislocalization, disrupted intracellular trafficking, instability, and altered function (Athanasiou et al., 2018). The Rho∆I256 mutation is a prevalent primary cause of adRP in a large British kindred as well as in patients from other Western European countries and China (Inglehearn et al., 1991; Artlich et al., 1992; Lorenz et al., 1993b; Li et al., 2010). So far, the precise mechanism and biochemical defects linking it to photoreceptor degeneration have remained unclear.
Our study reveals, for the first time, that a dominant-negative (DN) effect significantly contributes to Rho∆I256-associated retinal degeneration. RHO∆I256 accumulates in the endoplasmic reticulum (ER) as a substrate for VCP-dependent ER-associated degradation (ERAD). Like RHOP23H, RHO∆I256 forms perinuclear aggregates, hindering RHOWT from targeting the plasma membrane in cell models or reaching rod outer segments (ROS) in retinal explants. We show here that RHO∆I256 aggregates colocalize with VCP and undergo polyubiquitination before being retrotranslocated from the ER to the proteasome system. When inhibiting these processes pharmacologically, we observed decreased clearance of aggregates, indicating that both VCP and the proteasome play critical roles in the degradation of RHO∆I256 aggregates.
Biochemical analysis using HEK293 and COS-7 cells as models demonstrates that, unlike RHOWT and very similar to RHOP23H, RHO∆I256 protein mislocalizes in the perinuclear region of the cell cytosol, failing to target the cell membrane. Detergent-soluble and detergent-insoluble lysate fractions revealed a higher tendency for RHO∆I256 than RHOWT to form aggregates, including high molecular weight (HMW) aggregates exceeding 180 kDa, which is in line with previous studies (Sung et al., 1993). This again resembles aggregation patterns seen with RHOP23H (Illing et al., 2002; Griciuc et al., 2010a). Yet, there is a higher tendency for RHO∆I256 to form HMW aggregates than RHOP23H. This finding goes in line with clinical observations that patients with Rho∆I256 experience a more severe natural history of disease than those with RhoP23H (Inglehearn et al., 1991; Artlich et al., 1992; Lorenz et al., 1993a). This could be, at least in part, due to the fact that the deletion of one of the two isoleucine at position 255 or 256 within the sixth transmembrane domain of RHO shortens this intramembrane stretch significantly, affecting the subsequent 20 amino acids as well (Lorenz et al., 1993a).
Given that under normal conditions, RHO likely acts as a functional dimer, the impaired functionality of oligomeric aggregates is expected, hindering normal processes like G protein coupling, interaction with RHO kinase, and engagement with arrestin in the phototransduction cascade (Chabre and Le Maire, 2001; Whorton et al., 2007; Whorton et al., 2008). Nevertheless, the precise mechanisms of how these aggregates lead to cell death remain unknown. A dominant-negative (DN) effect of the mutant protein on the normal protein was reported to account for the dominance of other mutant Rho alleles in adRP inheritance (Illing et al., 2002; Saliba PMM et al., 2002; Wilson and Wensel, 2003; Griciuc, 2010). To explore whether RHO∆I256 has an effect on RHOWT, we conducted an experiment co-expressing EGFP-tagged Rho∆I256 with mcherry-tagged RhoWT in cells at equivalent dosage, mirroring conditions observed in RHO∆I256 heterozygous patients. The result revealed significant retention of RHOWT protein in the cell cytosol, colocalizing with intracellular RHO∆I256 aggregates. Similar outcomes were observed in cells co-transfected with RhoP23H and RhoWT, aligning with prior studies demonstrating that the RHOP23H mutation disrupts RHOWT processing, leading to protein inclusions within the cell and impeding RHOWT delivery to the plasma membrane (Illing et al., 2002). In contrast, co-expression of RhoWT-EGFP with RhoWT-mcherry did not result in aggregates with both RHOWT fusion proteins reaching the cell membrane. These findings confirm the dominant interference of both Rho∆I256 and RhoP23H into overall RHO trafficking, suggesting the physical interaction between mutant and wild-type protein.
We extended our study to a three-dimensional retinal explant culture system to better evaluate the DN effect in a more biologically and clinically relevant model. We expressed exogenous Rho plasmids in C57BL/6J (WT) retinal explants using the Magnetofection™ method (Sen et al., 2021a; Bassetto et al., 2021). In WT retinae transfected with mutant Rho∆I256-EGFP or RhoP23H-EGFP plasmids, fluorescence emitted by Rho∆I256-EGFP overlapped with that of RHOWT protein in the ONL, contrasting with RhoWT-EGFP-transfected retinae where both endogenous as well as exogenous RHOWT accurately localized to the OS. These results demonstrate that mutant RHO influences the expression or trafficking of endogenous RHOWT, confirming the DN effect of Rho∆I256 as well as that of RhoP23H in -adRP. Our findings on RhoP23H align with studies showing the interaction between RHOP23H aggregates and RHOWT through Förster resonance energy transfer (FRET) (Rajan and Kopito, 2005). Other publications affirm that while RHOP23H does aggregate; however, there is no physical interaction with RHOWT (Gragg and Park, 2018). Nonetheless, all studies, including ours, report colocalization between RHOP23H and RHOWT under microscopy, consistent with the notion that these different mutant RHOs hinder the correct trafficking of RHOWT to the plasma membrane. ER retention of aberrant RHO aggregates is one typical feature of class II adRP (Athanasiou et al., 2018). Rho∆I256 falls into this category, and we suggest it belongs to class II.
We found Rho∆I256 to form ER-associated aggregates colocalizing with VCP, ubiquitin, and the proteasome system, three important components of the ERAD process. RHOWT experiences repeated cycles of internalization-degradation (Satoh and Ready, 2005), and in our current study, RHOWT accumulated after VCP and proteasome inhibition, suggesting that RHOWT can also undergo ERAD degradation.
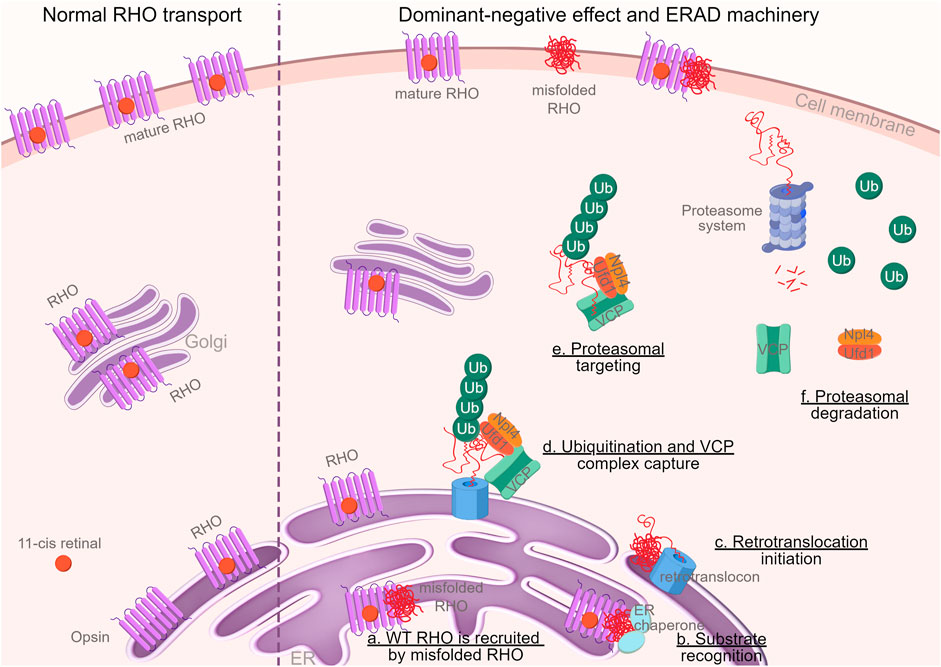
In conclusion, our research provides novel evidence supporting a dominant-negative role of the mutant allele in Rho∆I256-mediated retinitis pigmentosa. The observed formation and degradation of RHO∆I256 aggregates through the VCP-dependent ERAD pathway makes it a candidate for pharmacological therapy through VCP inhibition. The whole process was described in graphical abstract (Figure 8).
Figure 8. Graphical abstract. Schematic illustration of the dominant-negative mechanism of RHO∆I256 on RHOWT and the degradation of these mutant proteins in the ERAD machinery (By FigDraw, ID: TWOIA64640). Misfolded RHO gets withheld in the ER and traps RHOWT (a). Molecular chaperones, such as calnexin, calreticulin, and HSP70, identify misfolded proteins (b) and transport them to the ER retrotranslocation site, where the polypeptides are partially removed from the ER (c) and polyubiquitinated (d). Ubiquitination and VCP complex formation attract ubiquitin fusion degradation 1 (UFD1) and nuclear-protein localization 4 (NPL4) proteins to the retrotranslocon, which initiates the exit of the retrotranslocon from ER to cytoplasm (d). The complex retrotranslocates further into the cytoplasm (step d to e), where VCP-ATPase activity unfolds the protein (e). The unfolded VCP substrate is delivered to the proteasome for full degradation (f) (Vembar and Brodsky, 2008). Ubiquitin, VCP, and other cytosolic chaperones are then recycled.
However, further validation in animal model systems is crucial to confirm both the classification of Rho∆I256 as a class II adRP and a possible therapeutic option of targeting the VCP-dependent ERAD pathway for this form of adRP.
4 Materials and methods4.1 Primers design, cloning, and E. coli transformationPlasmids pEGFP-N1 encoding bovine Rho (WT or P23H) driven by the CMV promoter were provided by M. Cheetham (Saliba PMM et al., 2002). For the Rho∆I256 mutation, we designed the primers (IDT primer quest tool), forward primer: 5’-CCG CAT GGT CAT CAT GGT CAT CG-3’ and reverse primer: 5’-CAG GAA AGC GAT GAC CAT GAT GAC CAT GC-3’. Rho∆I256 was obtained and amplified by PCR using bovine RhoWT as template. To harvest single colonies that contained Rho∆I256, chemical transformation of competent E. coli was performed with 2.5 µL of PCR reaction product added into 50 µL of cells. After that, plasmid DNA was prepared from small-scale (1.5 mL, miniprep) bacteria cultures using QIAprep Spin Plasmid Miniprep Kit or from large-scale (50 mL) overnight cultures using the PureYield Plasmid Midiprep System. (Notes: Isoleucine residues at codons 255 and 256 are conserved in bovine and human Rho. So, we designed the deletion of isoleucine at codon 256 based on the wild-type bovine Rho backbone in the pEGFP vector and applied it in our study.)
4.2 Cell culture and transfectionsHEK293 human embryonic kidney cells (CRL-1573, ATCC—Global Bioresource Center) and COS-7 African green monkey kidney fibroblast-like cells (CRL-1651, ATCC—Global Bioresource Center) were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (Thermofisher Scientific). In addition, cells were transiently transfected with 1 µg of Rho expression plasmids using TransIT-LT1 transfection reagent (MIR2300, Mirus). HEK293 and COS-7 cells were cultured on the Poly-D-lysine (P6407, Sigma) coating 24 well plates with autoclaved glass coverslips (1.2 cm diameter) for immunostaining or 6 well plates without coverslips for Western blot. Cells were transiently transfected with Rho-EGFP constructs or co-transfected with Rho-EGFP and RhoWT-mcherry after 80% of cell confluency and incubated for 48 h.
4.3 AnimalsC57BL/6J mice were obtained from Jackson Laboratory (JAX stock #000664, RRID: IMSR_JAX:000664) and housed in the animal facility of the Tübingen Institute for Ophthalmic Research under standard white cyclic lighting, had free access to food and water, and were used irrespective of gender. All procedures were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) declaration for the use of animals in ophthalmic and vision research and the law on animal protection issued by the German Federal Government (Tierschutzgesetz) and were approved by the institutional animal welfare office of the University of Tübingen. All efforts were made to minimize the number of animals used and their suffering.
4.4 Immunofluorescence and microscopyCells were fixed and stained 48 h after transfection, as described before (Mendes and Cheetham, 2008). Samples were incubated overnight at 4°C with the primary antibodies (calnexin: C5C9, Rabbit monoclonal, 1:100, Cell signaling; VCP: mouse monoclonal, 1:200, Invitrogen; ubiquitin: sc8017, mouse monoclonal, 1:50, Santa Cruz; or proteasome subunit 20S: PSMB5, rabbit polyclonal, 1:100, Invitrogen followed by incubation with secondary antibodies (IgG Alexa Fluor™ 568 or 660 dye-conjugated goat anti-mouse or anti-rabbit respectively, Molecular Probes, Inc. Eugene, United States of America). Negative controls were carried out by omitting the primary antibody. To measure RHO immunofluorescence intensity, the whole cell body of HEK293 cells (n ≥ 40) and COS-7 cells (n ≥ 20) of three seperate experimental replicates were carefully contoured, and the fluorescence intensity value was automatically measured by ZEN software. Colocalization analysis using Pearson’s correlation coefficient between RHO and ERAD markers was performed using ZEN software. We select at least 6 distinct regions on the cell coverslip for data analysis. All images were captured and analyzed using a Zeiss Axio Imager Z1 ApoTome microscope, AxioCam MRm camera, and Zeiss Zen 2.3 software at 20X or ×63 magnification.
4.5 VCP- and proteasome inhibition assayHEK293 cells were incubated with 5 µM proteasome inhibitor MG132 (J63250, Alfa Ascar, Thermo Fischer Scientific) or 2 µM VCP inhibitor, ML240 (5,153, Tocris) dissolved in 0.1% DMSO for 6 h after 48 h of transfection. For each inhibitor, 0.1% DMSO was applied as vehicle control. Cell lysates were then harvested and analyzed by Western blot.
4.6 Western blotHEK293 cells transfected with Rho-EGFP were harvested after 48 h of incubation and then lysed at 4°C in 0.5% Nonident P40 (NP40, Roche) lysis buffer with 1% phosphatase inhibitor cocktail 2 and 3 (PI2 and PI3, Sigma), 2% protease inhibitor complex (PIC, Roche) in TBS. Lysates were centrifuged at 16,000 g for 15 min at 4°C. For detergent-insoluble fractions, pellets were solubilized in 1% SDS in PBS at RT, and lysis buffer was added. The pellets were then sonicated at 4°C, and the fraction mixtures were incubated 30 min and then centrifuged at 100 g for 10 min, both at 4°C (Griciuc et al., 2010b). For detergent-soluble/-insoluble samples, protein concentration was determined by Bradford assay kits. Cell lysates were normalized for total protein, and 7.5 µg (RHO protein in detergent-soluble), 10 µg (RHO in detergent-insoluble fractions), or 20 µg (Ub protein or RHO protein after VCP inhibition) total protein was prepared and mixed with 1x Laemmli sample buffer before being separated by 8%–16% Tris-Glycine precast gels (Invitrogen) and electroblotted onto PVDF membranes. Immunoblotting was performed according to standard protocols using the as primary antibodies: anti-RHO (1D4, MAB5316, 1:300, Sigma), anti-VCP (MA3-004 1:1,000, Invitrogen), anti-ubiquitin (sc8017, 1:500, Santa Cruz), and anti-β-actin (3700S 1:2000, cell signaling). Afterward, horseradish peroxidase-coupled (HRP) secondary antibodies were applied (1:2000, cell signaling). The ECL Plus chemiluminescent and chemifluorescent HRP detection reagent (Thermo Scientific™ Pierce™ ECL) was used according to the manufacturer’s instructions. Band intensity was quantified by ImageJ software, and relative protein expression levels were determined based on the WT or untreated control group.
4.7 Organotypic culture preparation and classic magnetofectionIn vitro retina cultures were prepared according to published protocols (Arango-Gonzalez et al., 2010). PN9 animals were sacrificed, and the eyeballs were removed and collected in a serum-free R16 culture medium (07490743A, Invitrogen Life Technologies). The eyes were pretreated with 0.12% Proteinase K (0219350490, MP Biomedicals) for 15 min at 37°C in R16 medium. After enzymatic digestion of the eyeball, the retina and the attached pigment epithelium were dissected, and four radial cuts were made to flatten it. Retinas were placed on culture plate inserts (CLS3412, Corning Life Sciences) with the ganglion cell layer uppermost. The retinal explants were cultured in 1 mL serum-free culture medium consisting of Neurobasal A (21103049, Thermo Fischer Scientific) supplemented with 2% B-27 supplement (17504044, Thermo Fischer Scientific), 1% N2 supplement (17502048, Thermo Fischer Scientific), 1% penicillin solution (15140-122, Thermo Fischer Scientific), and 0.4% GlutaMax (35050061, Thermo Fischer Scientific) and maintained at 37°C in a humidified atmosphere with 5% CO2. At the same time, 2 µg EGFP-tagged RhoWT, RhoP23H, Rho∆I256 plasmids, and 1.5 µL magnetic nanoparticles (XPMag Explant Transfection Reagent, OZBiosciences) were complexed for 30 min in Neurobasal-A medium, to a total volume of 10 µL. The solution was incubated at room temperature for 30 min to allow the cationic magnetic nanoparticles to bind to the negatively charged DNA plasmids. After complexation, the magnetic complexes were administered to the top of each retina (on the ganglion layer side). The super-magnetic plate was immediately placed under the 6-well plate. Then, the set well plate/magnet was kept in the incubator for 1 h, to allow the pull down of the complexes through the retinae. After removal of the magnet, retinae in the 6-well plates remained incubated at 37°C in 5% CO2 for 3 days before fixation in 4% paraformaldehyde.
4.8 Preparation and immunohistochemistry on retinal sectionsThe explants were immersed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) for 45 min at RT, followed by cryoprotection in graded sucrose solutions (10%, 20%, 30%) and embedded in cryomatrix (Tissue-Tek® OCT Compound, VWR). Radial sections (14 µm) were collected, air-dried, and stored at −20°C. Retina sections were incubated overnight at 4°C with rhodopsin primary antibody (1D4, MAB5316, 1:300, Sigma) followed by incubation with secondary antibody (Alexa Fluor™ 568 dye-conjugated goat anti-mouse IgG). DAPI (D9542, Sigma) was used as a nuclear counterstain. Finally, the slides were mounted with Fluoromount-G Mounting Medium (17984-25, EMS).
4.9 Statistical analysesMann-Whitney-U-test analysis was used to examine RHO MFI that did not match Gaussian distribution; while, Pearson’s correlation coefficient and protein gray levels that were consistent with normal distribution were analyzed using one-way ANOVA followed by Bonferroni’s post hoc comparisons tests in GraphPad Prism 9 software. Data were presented as mean ± standard error of the mean (SEM). Differences were considered significant and indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statementEthical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by all procedures were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) declaration for the use of animals in ophthalmic and vision research and the law on animal protection issued by the German Federal Government (Tierschutzgesetz) and were approved by the institutional animal welfare office of the University of Tübingen. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributionsBC: Conceptualization, Writing–original draft, Visualization, Methodology, Investigation, Formal Analysis, Data curation. JD: Methodology, Writing–review and editing, Investigation, Data curation. MS: Methodology, Writing–review and editing, Investigation. TB: Conceptualization, Writing–review and editing, Methodology. TL: Writing–review and editing, Methodology. EK: Writing–review and editing, Project administration, Writing–original draft, Conceptualization. BA-G: Writing–review and editing, Visualization, Investigation, Formal Analysis, Writing–original draft, Supervision, Project administration, Funding acquisition, Conceptualization. MU: Writing–review and editing, Resources, Investigation, Formal Analysis, Writing–original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Tistou and Charlotte Kerstan Foundation (to MU and BA-G), Fighting Blindness Canada (FBC)—2022–23 Transformative Research Award (to MU and BA-G) and by institutional funds to MU.
AcknowledgmentsThe animal husbandry personnel at the Universitätsklinikum Tübingen and Norman Rieger are acknowledged for providing animal care. We acknowledge support from the Open Access Publishing Fund of the University of Tübingen, and gratefully acknowledge the support from the China Scholarship Council (grant number 201808080183).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2024.1369000/full#supplementary-material
Ref
留言 (0)