
記住我




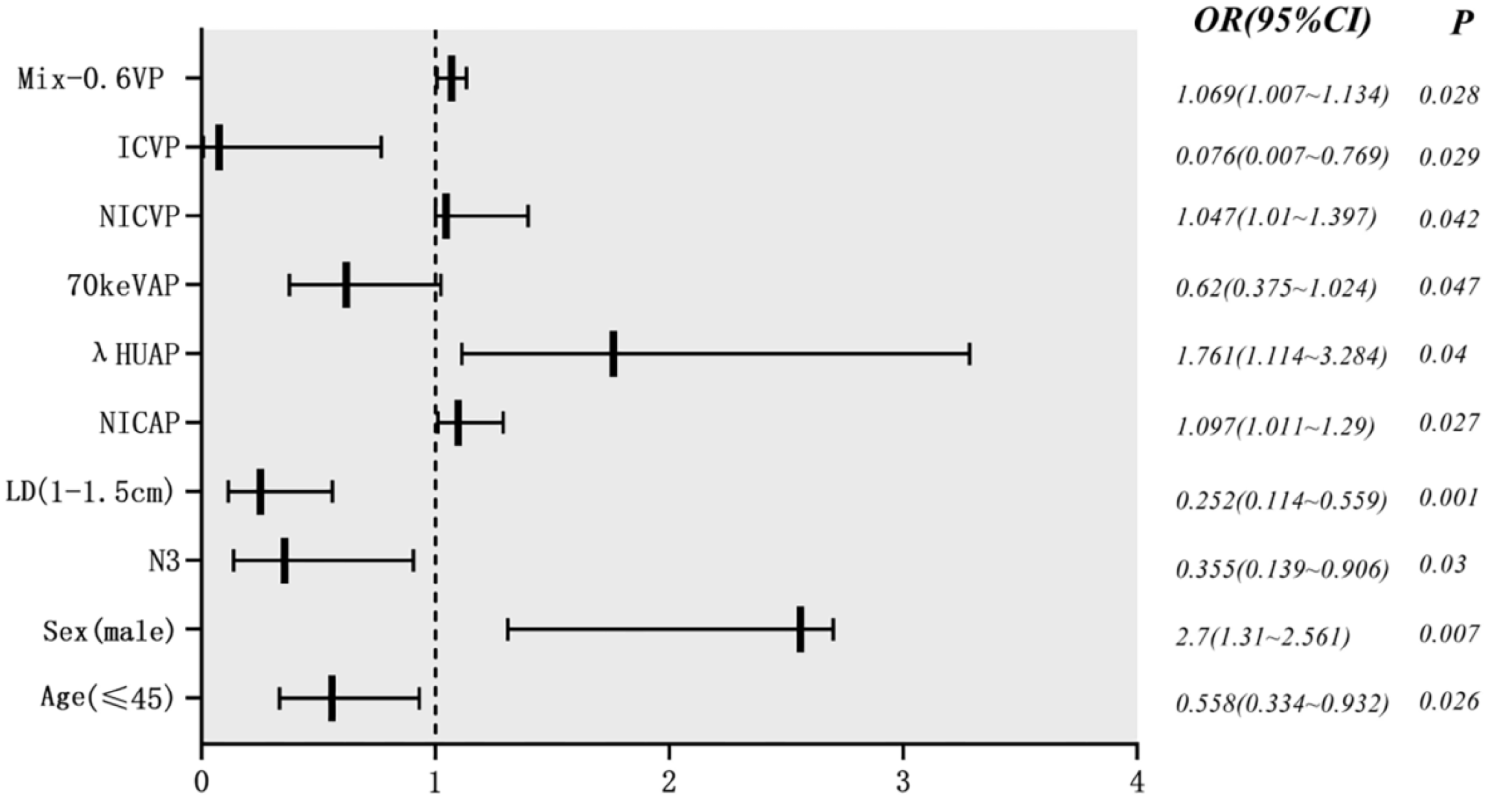


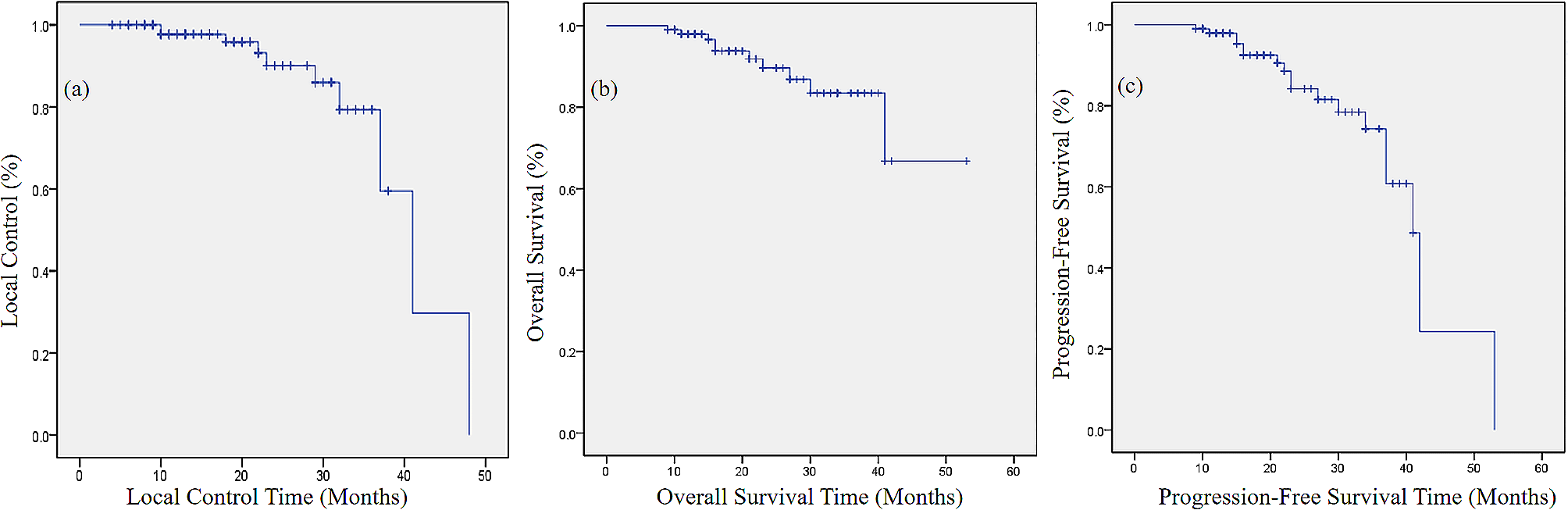
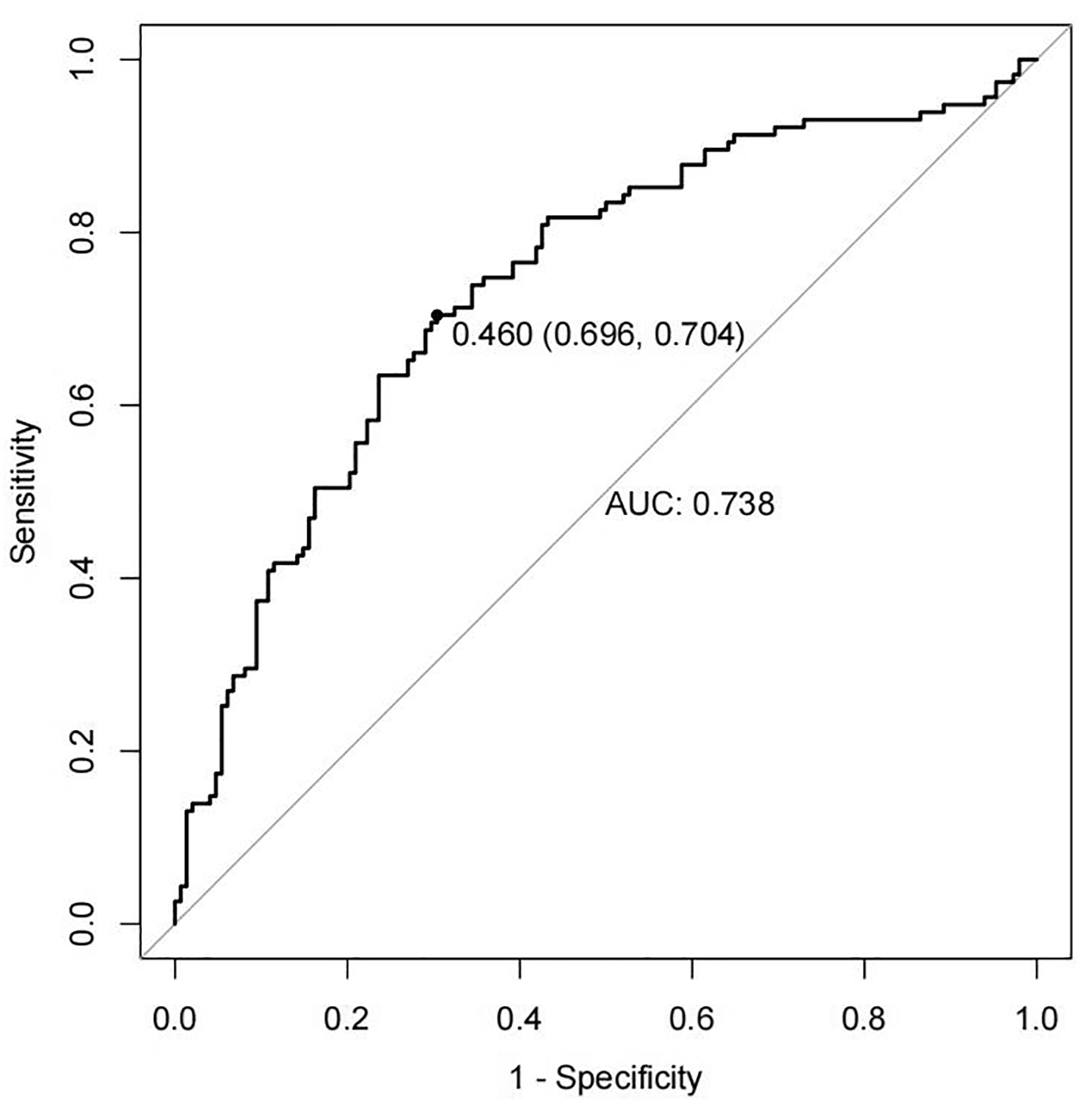
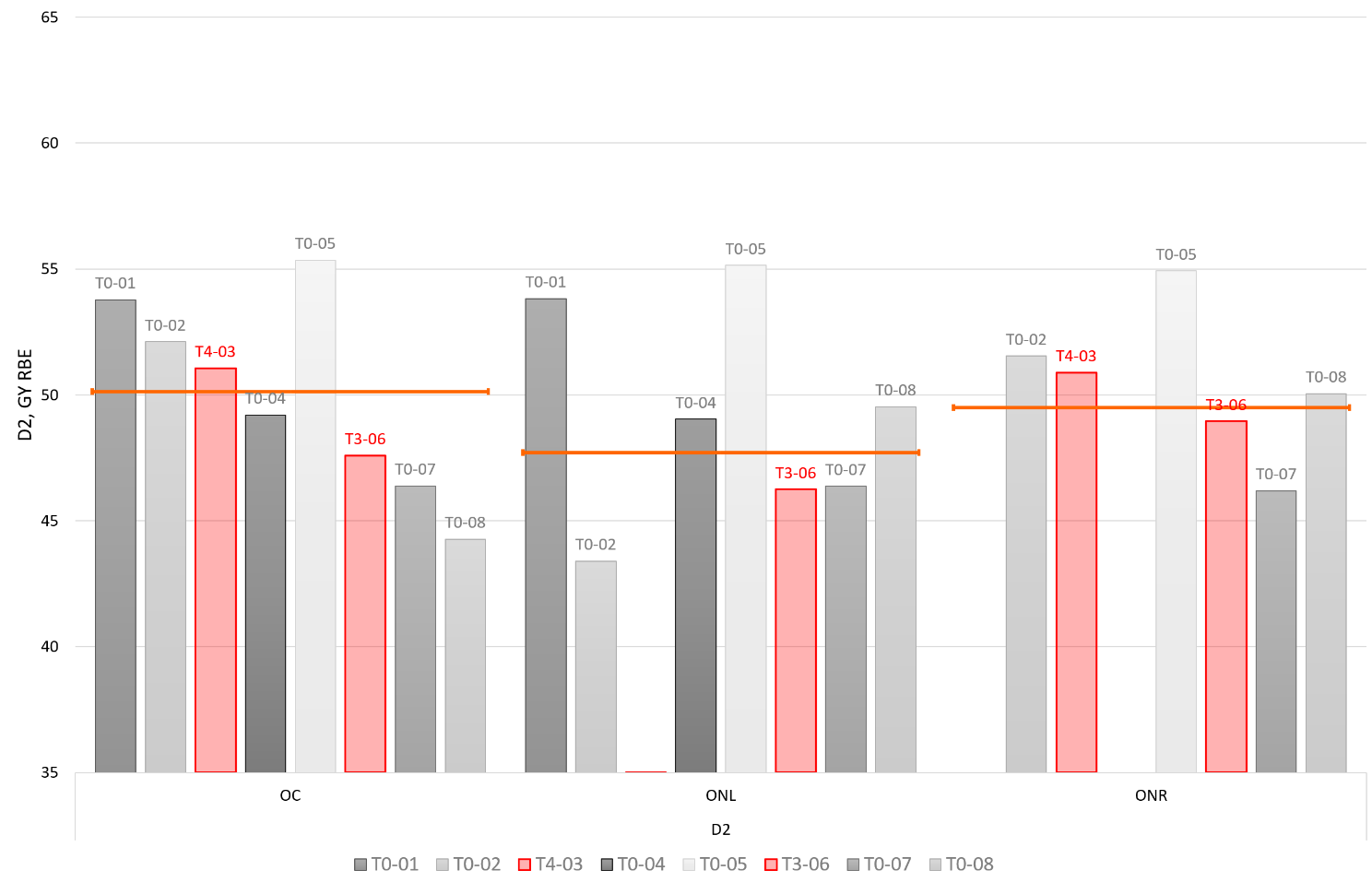
This study titled “PROspective phase II trial of preoperative hypofractionated protoN therapy for extremity and Truncal soft tissue sarcOma (PRONTO)” is a prospective, single arm, phase II clinical trial designed to assess the safety and efficacy of preoperative, hypofractionated PBT for patients with extremity and truncal STS planned for surgical resection. We plan to accrue 40 patients who meet all inclusion and no exclusion criteria, as outlined in Table 2. The trial was activated April 2024, and we anticipate accrual to begin May 2024. With an anticipated accrual of 24–36 months for all 40 patients, we aim to complete accrual by late 2026 or early 2027.
Table 2 Inclusion and exclusion criteriaTrial organizationThis trial was designed by the Departments of Radiation Oncology and Molecular Radiation Sciences, Oncology, Orthopaedic Surgery, and Physical Medicine and Rehabilitation of Johns Hopkins University School of Medicine. It is carried out by the Johns Hopkins Proton Therapy Center together with the Department of Radiation Oncology and Molecular Radiation Sciences at Johns Hopkins University School of Medicine. It is an investigator-initiated trial.
InvestigatorsPatients will be recruited by the Departments of Radiation Oncology and Molecular Radiation Sciences and Orthopaedic Surgery of Johns Hopkins University School of Medicine. All investigators cooperating in this trial are experienced oncologists from the fields of radiation oncology and orthopaedic surgery.
Ethical and legal considerationsThe study protocol was approved by the Clinical Research Review Committee and the Institutional Review Boards of Johns Hopkins University School of Medicine (IRB00335181). The trial is carried out by adhering to local legal and regulatory requirements. All patients will sign informed consent before enrollment on trial after the nature, scope, and potential consequences of participation are explained by a physician.
Study objectives and endpointsThe primary outcome is the rate of major wound complications occurring within 90 days after surgery, as defined according to the National Cancer Institute of Canada Sarcoma2 (NCIC-SR2) Multicenter Trial [7, 8]. This includes “secondary operation under general or regional anaesthesia for wound repair (debridement, operative drainage, and secondary wound closure including rotationplasty, free flaps, or skin grafts), or wound management without secondary operation…[including] an invasive procedure without general or regional anaesthesia (mainly aspiration of seroma), readmission for wound care such as intravenous antibiotics, or persistent deep packing for 120 days or longer.” Secondary objectives include safety and tolerability (acute grade ≥ 3 adverse events based on National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0 [CTCAE v5.0]), 1- and 2-year LRFS and distant metastasis-free survival (DMFS) rates, incidence of CTCAE v5.0 late grade ≥ 2 radiation toxicity (fibrosis, lymphedema, or joint stiffness), functional outcomes using the Musculoskeletal Tumor Rating Scale (MSTS) and Toronto Extremity Salvage Score (TESS), quality of life assessed via Functional Assessment of Cancer Therapy-General (FACT-G) forms, and pathologic response (complete response, positive margins, and percentage necrosis in comparison with pre-treatment biopsy when available).
Pretreatment evaluationTable 3 outlines time flow for all work up, enrollment, interventions, and assessments.
Table 3 Time flow for all work up, enrollment, interventions, and assessmentsInitial work up includes clinical evaluation, staging computed tomography (CT) and/or magnetic resonance imaging (MRI) of the primary site, chest CT, histologic confirmation of STS, and determination of eligibility and resectability by clinical assessment and laboratory studies.
Treatment assignment and scheduleAll eligible patients who provide informed consent are registered and follow work-up and treatment as outlined in Fig. 2.
Fig. 2
Flow chart outlining trial schema, study procedures, follow-up, and planned analysis. Abbreviations: STS, soft tissue sarcoma; yrs, years; ECOG PS, Eastern Cooperative Oncology Group Performance Status; GyE, Gy radiobiologic equivalent; PBT, proton beam therapy; CTV, clinical target volume; OTV, on treatment visit; CTCAE, common terminology criteria for determining adverse events version 5.0; q3-6mo x2yrs, every 3–6 months for 2 years; CT, computed tomography; MRI, magnetic resonance imaging; SOC, standard of care
Treatment detailsEnrolled patients undergo CT simulation. Due to the variety of sites in which STS may occur and the mobility of extremities, careful consideration of immobilization is required prior to simulation. Immobilization must be reproducible to a high degree. Examples include the use of large vac loc or body fix bags for lower extremity immobilization with the addition of aquaplast mold to immobilize the foot and/or knee.
After completing simulation, patients begin PBT within 1–3 weeks from their simulation, treating with 5 fractions given every other weekday, to a total dose of 30 Gy radiobiological equivalent for a total of 10–12 calendar days from treatment start to completion. Clinical target volumes include gross tumor with a 3 cm margin longitudinally and 1.5 cm margin radially excluding natural barriers of spread, in addition to any surrounding edema seen on T2 weighted MRI, cropped 0.3 cm from the skin. In place of the planning target volumes (PTVs) typically used when treating with photon radiation, robust optimization of PBT is used to account for setup and range uncertainties. Target and organ at risk planning goals are listed in Table 4A, B.
Table 4 (A) Target dose and coverage parameters, (B) Normal tissue constraintsPatients undergo daily kilovoltage x-ray and cone beam CT (CBCT) before daily PBT to assist with treatment set-up. Quality assurance (QA) verification CT +/− MRI are performed if feasible within the first 2 fractions and repeated during the treatment course as needed.
Oncologic surgical resection occurs within 2–12 weeks of completing PBT. Patients will be followed in the postoperative setting according to standard of care surveillance for STS, as outlined in Fig. 2.
Outcomes measured and follow-upMedical and demographic details, performance status (Eastern Cooperative Group Performance Status scale), laboratory studies, and imaging are captured at baseline. CTCAE v5.0 toxicity, functional status using MSTS and TESS forms, and quality of life assessed by FACT-G forms are captured at baseline, during PBT, after PBT but prior to surgery, within 3 months after surgery, and every 3–6 months thereafter. Pathologic response is evaluated using rates of pathologic complete response, positive margins, and percentage necrosis in comparison with pre-treatment biopsy. Postoperative wound complications are assessed after surgery at 2 weeks, 4–6 weeks, and every 3–6 months thereafter. LRFS is assessed clinically and by postoperative MRI +/− CT of the primary site at 3 and 6 months after surgery, CT and/or MRI every 3–6 months for the first 2 years, and every 6–12 months thereafter. DMFS is assessed via chest CT or X-ray at 3 months after surgery, every 3–6 months for the first 2 years, and every 6–12 months thereafter. Patients will be followed for a minimum of 2 years on study.
Statistical considerationsThe primary outcome, rate of major wound complications, will be estimated as the number of major wound complications occurring within 90 days after surgery during the study period divided by the number of evaluable patients who have completed PBT and surgery. A sample size of 36 evaluable patients will allow us to obtain a two-sided 90% exact confidence interval with a width less than 0.3 if the incidence is between 30 and 60%, based on those reported in multiple prospective trials in patients receiving preoperative, hypofractionated photon-based EBRT followed by surgical resection for STS [13, 24].
Guarding against the potential that 10% of patients may be not evaluable, we plan to accrue 40 patients in total in order to provide 36 evaluable patients. Patients will be considered evaluable as long as they have received 1 fraction of hypofractionated PBT and completed surgery.
Safety of hypofractionated PBT in isolation will be analyzed by calculating the incidence of CTCAE v5.0 grade ≥ 3 adverse events occurring between receipt of the first fraction of PBT and the day of surgery. Specific adverse events will be reported as frequencies. Patient or cancer characteristics associated with acute PBT-related toxicity will be explored with logistic regression models provided a sufficient number of events. Tolerability will be defined and reported as the frequency of patients who stop treatment with PBT due to an adverse event.
LRFS and DMFS will be reported at 1- and 2-years post-enrollment based upon estimates produced using Kaplan–Meier methods. Patient and tumor characteristics associated with LRFS and DMFS will be explored using Cox proportional hazards models. A sensitivity analysis will be performed including patients who completed all 5 fractions of their hypofractionated PBT if this does not apply to the entire population.
Early stopping rulesBased on existing literature, we assume the rate of major wound complication within 90 days after surgery when treated with preoperative photon RT is about 25–30%. Therefore, to minimize risks, safety will be monitored by a Bayesian stopping rule for the rate of major wound complications greater than 60%. Table 5 summarizes the continuous stopping rule for the 36 evaluable patients, evaluated in cohorts of 3 patients, starting from 6th evaluable patient.
Table 5 Stopping rule for safetyAt any time if the stopping criterion is met, accrual to the trial will be temporarily suspended, and the principal investigators and study team will review the toxicity data to recommend modification or termination of the trial.
留言 (0)