
記住我






The administration of vaginal progesterone (VP) to prevent spontaneous preterm birth in women with a sonographic short cervix has become part of the standard practice in the United States and has been recommended by professional societies, such as the American College of Obstetricians and Gynecologists, the Society of Maternal-Fetal Medicine, the National Institute for Health and Care Excellence, and the International Federation of Gynecology and Obstetrics. The recommendation is based upon the results of randomized clinical trials (RCTs) and systematic reviews and meta-analyses, as well as cost-effectiveness analyses and implementation research. The PREGNANT trial made an important contribution toward the implementation of VP to prevent preterm birth.1,2 This article will review the scientific rationale for using progesterone for this indication, as well as the study design, results, and impact of the PREGNANT trial in obstetrics.
Progesterone: A Key Hormone for Pregnancy MaintenanceProgesterone (from pro “for” and gest “pregnancy”), a natural steroid hormone essential for pregnancy maintenance, is produced by the corpus luteum3–5 until the seventh to the ninth week of gestation after which time the placenta becomes the main source, a transition known as “the luteal-placental shift.”6 Removal of the corpus luteum before the seventh week results in spontaneous abortion, but this can be prevented by the administration of progesterone.7–9 Luteectomy after 9 weeks does not result in pregnancy loss. The plasma progesterone concentration increases during pregnancy from 40 ng/mL in the first trimester to 160 ng/mL in the third trimester.10 The placenta at term produces ~250 mg of progesterone per day, of which 90% is secreted to the maternal circulation and 10% into the fetal circulation.11
Progesterone has multiple biological effects, but it was discovered because of its effect on the endometrium. During the normal menstrual cycle, progesterone transforms the endometrium from proliferative during the follicular phase of the menstrual cycle to secretory during the luteal phase.12,13 The primary effect of progesterone is the transformation of endometrial stem cells into specialized secretory “decidual cells” that provide nourishment and a receptive microenvironment for embryo implantation and placental development.12,13 In the absence of blastocyst implantation, the corpus luteum regresses, which leads to decreased production of progesterone, “a progesterone withdrawal,” and menstruation.12,13 The molecular and cellular events responsible for menstruation are similar to those involved in parturition. Both are considered inflammatory processes14–16 involving increased production of (1) inflammatory mediators, such as cytokines/chemokines,17,18 interleukin (IL)-8, and IL-1,19,20 (2) cyclooxygenase-2 and prostaglandins,21,22 (3) matrix metalloproteinases,23–25 and (4) the transcription factor nuclear factor-kappa B (NF-κB).26–28
Progesterone production by the placenta after the luteo-placental shift maintains pregnancy by preventing activation of the common pathway of parturition.1 In other words, this hormone induces myometrium quiescence, prevents cervical ripening, and supports the decidua and chorioamniotic membranes.1
Conceptually, parturition can be considered a “delayed menstruation.”29,30 The delay occurs in the production of progesterone by the placenta. In most mammalian species, there is a drop in circulating progesterone concentration before the onset of labor.31 However, such a decrease does not occur in humans, non-human primates, and some species, such as guinea pigs.32–35 Yet, there is considerable evidence that a decline in progesterone action occurs before labor in primates, and this has been attributed to a functional progesterone withdrawal.36,37 This term refers to a decline in progesterone action (as assessed by changes in progesterone-targeted organs) in the absence of a decrease in the concentration of the hormone in peripheral blood.
A Decline in Progesterone Action as a Cause of Preterm LaborSpontaneous preterm labor is a syndrome caused by multiple etiological processes including intra-amniotic infection, sterile intra-amniotic inflammation, uterine over-distention, and a breakdown of maternal-fetal tolerance.38–40 In a subset of patients, a decline in progesterone action is thought to cause a short cervix and, subsequently, preterm labor.1,41 The administration of VP is predicated on the hypothesis that this hormone can prevent progression toward preterm labor.1,41
Most of the studies about the effect of progesterone on the uterus have focused on myometrial contractility. However, there is a considerable body of evidence that progesterone has powerful effects on the uterine cervix. Such evidence includes (1) administration of antiprogestins to women in the first, second, and third trimesters of pregnancy to induce cervical ripening42–47 and (2) progesterone-receptor antagonists, such as mifepristone (RU486) or onapristone, also causes cervical ripening in pregnant guinea pigs,48,49 old-world monkeys,50 and Tupaja belangeri.51 Such a response increases with advancing gestational age,47 and the effects on the cervix are not always accompanied by changes in myometrial activity.47 Indeed, Stys et al52 demonstrated a functional dissociation between the effects of progesterone in the myometrium and in the cervix. In animals,48,50 as well as in humans,51 antiprogestin treatment induces cervical ripening but not necessarily labor or abortion. Labor may not begin at all or may be delayed by days or weeks in humans after cervical ripening has been accomplished.47 Collectively, these observations suggest that the uterine cervix is a major target organ for progesterone.
How does a progesterone blockade induce cervical ripening? A decline in progesterone action is thought to cause ripening by inducing a sterile inflammatory process (leukocyte infiltration and production of chemokines).53 The cervix is fundamentally an extracellular connective tissue organ, and inflammation induces the degradation of collagen and other changes that make the uterus soft and compliant. A role for the transcription factor NF-κB has been proposed because this molecule induces inflammation through the effects of inflammatory cytokines such as IL-1β,26,54–61 tumor necrosis factor-α,62–64 IL-8,65,66 matrix-degrading enzymes,67–69 nitric oxide,46,70,71 and prostaglandins.66 It is noteworthy that NF-κB can oppose progesterone action.27,55,61,72–75 Thus, NFκB provides a link between inflammation, a decline in progesterone action, and cervical ripening.
Progestogens: Vaginal Progesterone and 17α-Hydroxyprogesterone CaproateProgestogens can be classified as natural or synthetic.76–79 Natural compounds are those with chemical structures similar to those produced by living organisms. By contrast, synthetic progestogens (or progestins) are compounds generated in the laboratory whose structures have been modified and do not correspond to a naturally occurring steroid. Progesterone is a natural progestogen; 17α-hydroxyprogesterone caproate (17-OHPC) is synthetic. These 2 compounds have different effects on the uterine cervix. VP has been shown to reduce the rate of cervical shortening in patients with a history of preterm birth or premature cervical shortening.80 By contrast, 17-OHPC does not have an effect on cervical length in patients with a history of ≥1 preterm births.81 In pregnant mice, neither VP nor 17-OHPC changed myometrial gene expression substantially.82 However, in the cervix, there was a difference in the 2 compounds. VP up-regulated the expression of defensin-1 (a natural antimicrobial peptide), but 17-OHPC had no detectable effects on gene expression.82 A comprehensive study comparing the local effects of VP and 17-OHPC in pregnant mice showed that VP, but not 17-OHPC, had anti-inflammatory effects at the maternal-fetal interface. For example, VP reduced the abundance of neutrophils and monocytes expressing matrix metalloproteinase-9 in cervical tissue while 17-OHPC increased their abundance. Moreover, VP pretreatment protected against endotoxin-induced preterm delivery (for details, Furcron et al83).
Progestogens to Prevent Preterm Labor and Birth: Randomized Trials That Led to the Conceptualization of the Pregnant TrialThe use of progestogens to prevent spontaneous preterm birth was pioneered by Emile Papiernik84 in France and published in the 1970s. Subsequently, other investigators tested the effect of progestogens in the prevention of preterm birth or recurrent miscarriage. These studies yielded contradictory results as did meta-analyses of such trials.85–87 The meta-analysis reported by Goldstein et al85 did not show evidence of a beneficial effect on preterm birth. By contrast, Keirse86 focused on studies of 17-OHPC and reported in 1990 that 17-OHPC reduced the rate of preterm labor, preterm birth, and birthweight below 2500 g.
In 2003, the results of 2 RCTs rekindled the interest in progestogens to prevent preterm birth. da Fonseca et al88 reported that VP reduced the rate of preterm birth in patients at risk (clinical histories, such as prior preterm birth, uterine malformations, or a suspected incompetent cervix); preterm birth <37 weeks was 28.5% in the placebo group and 13.8% in the progesterone group (P = 0.03) and <34 weeks was 18.6% in the placebo group versus 2.8% in the progesterone group (P = 0.002). The second trial, reported by Meis et al,89 used 17-OHPC (250 mg intramuscular between 16 wk and 36 wk of gestation) and found a substantial decrease in the rate of preterm delivery <37 weeks [placebo, 54.9% vs 17-OHPC, 36.3%; relative ratio (RR): 0.66, 95% CI: 0.54-0.81] and <35 weeks (placebo, 30.7% vs 17–OHPC, 20.6%, RR: 0.67, 95% CI: 0.48–0.93). Both publications were accompanied by editorials that further sparked the interest in progestogens.
A key study on the effect of VP was reported by O’Brien et al.90 In a large RCT, patients with a history of previous spontaneous preterm birth were allocated to receive VP (90 mg daily) in a bio-adhesive formulation/gel or placebo. VP did not decrease the rate of spontaneous preterm birth ≤32, 35, or 37 weeks. The results of this study conflicted with the previous report by Fonseca et al.91 Several explanations were proposed to account for the difference, including the dose of progesterone and the criteria for patient selection.
DeFranco et al92 examined the effect of VP on pregnancy outcome (preterm birth and infant outcome) as a function of the cervical length of patients enrolled in the study by O’Brien et al.90 Patients eligible to participate in the trial by O’Brien et al90 had undergone cervical examination at the time of enrollment. DeFranco et al92 reported in patients with a cervical length <28 mm that the frequency of preterm delivery ≤32 weeks and the frequency of neonatal intensive care unit (NICU) admission were lower in patients who had received progesterone versus placebo. This analysis suggested that VP may reduce preterm birth and improve infant outcomes in properly selected patients. However, this conclusion was derived from a secondary analysis and, therefore, considered to be hypothesis-generating.
A step forward was the results of an RCT by the Fetal Medicine Foundation in which VP reduced the rate of preterm birth in women with a sonographic short cervix (≤15 mm). This study enrolled women with a short cervix between 20 weeks and 25 weeks of gestation and allocated patients to receive daily VP (200 mg) or placebo from 24 to 32 weeks of gestation. The rate of spontaneous preterm delivery <34 weeks (primary end point for the trial) was significantly lower in the VP group compared to the placebo group [19.2% (24/125) vs 34.4% (43/125), P = 0.007]. However, there was no difference in the rate of neonatal morbidity.91
The report by O’Brien et al90 indicated that VP was not effective in reducing preterm birth in patients with a prior history. However, VP was effective in the subset of patients who had a short cervix. This was consistent with the report of the Fetal Medicine Foundation. A key issue was whether VP reduced not only preterm birth but also neonatal morbidity, which was the impetus for the PREGNANT Trial.
The PREGNANT TrialThis section will review the key features of the design of the PREGNANT trial, followed by the results.
OBJECTIVEThis study was undertaken to determine the efficacy and safety of VP gel in reducing the rate of preterm birth before 33 weeks in asymptomatic women with a singleton gestation and a mid-trimester sonographic short cervix.
STUDY DESIGNThe trial was a phase 3, randomized, placebo-controlled, double-masked, parallel-group, multicenter, international trial. The study was conducted from March 2008 to November 2010.
SCREENING AND ELIGIBILITY CRITERIAWomen between 19 + 0 and 23 + 6 weeks of gestation were eligible for screening. During a screening visit, cervical length and gestational age were determined. Women were eligible for the study if they met the following criteria: (1) singleton gestation, (2) gestational age between 19 + 0 and 23 + 6 weeks, (3) transvaginal sonographic cervical length between 10 mm and 20 mm, and (4) asymptomatic, that is, without signs or symptoms of preterm labor. Patients were allocated randomly to receive VP gel 90 mg/d or placebo beginning at 20 to 23 + 6 weeks.
EXCLUSION CRITERIAThe exclusion criteria are as follows: (1) Planned cerclage, (2) acute cervical dilation, (3) allergic reaction to progesterone, (4) current or recent progestogen treatment within the previous four weeks, (5) major fetal anomaly or known chromosomal abnormality, (6) uterine anatomic malformation (eg, bicornuate uterus, septate uterus), (7) vaginal bleeding, (8) known or suspected clinical chorioamnionitis, or (9) chronic medical conditions that would interfere with study participation or evaluation of the treatment.
SONOGRAPHIC CERVICAL LENGTH DETERMINATIONCervical length was measured with transvaginal sonography. Sonographers involved in the study were required to participate in a training program and to obtain certification before screening patients for the trial. The sonographic images of patients enrolled in the trial were reviewed by a central sonologist for quality assurance. Patients were not scheduled to have follow-up examinations unless clinically indicated.
RANDOMIZATIONPatients were randomly allocated in a 1:1 ratio (VP:placebo), and this was accomplished by using a centralized interactive voice response system. Randomization was stratified according to (1) center and (2) risk strata (previous preterm birth between 20 wk and 35 wk of gestation or no previous preterm birth) using a permuted block strategy with a block size of four (ie, 2 placebo and 2 VP gel).
Three approaches were used to maximize concealment allocation. First, drug kits at each study site were numbered independently from the treatment assignments in the randomization blocks to avoid identification of dispensing patterns. Second, the interactive voice response system (upon generating a treatment assignment for a new patient) specified which kit number was to be dispensed to the patient. Third, the study drug packaging, applicators, and their contents (VP and placebo) were identical in appearance.
STUDY DRUGSThe drug required for the entire treatment of a randomized patient was included in the drug kits to be assigned to each patient at each study visit to prevent dispensing errors. Treatment was to be initiated between 20 + 0 weeks and 23 + 6 weeks of gestation. Women self-administered the study drug once daily in the morning.
COMPLIANCEStudy participants were asked to return to the study center every 2 weeks. During each visit, patients were interviewed to determine the occurrence of adverse events, use of concomitant medications, and compliance with the study drug. Women were asked to return unused study drugs from the previous 2 weeks, and the determination of compliance was based on the amount of study drugs not used.
VAGINAL PROGESTERONE AND PLACEBO ADMINISTRATION AND DISCONTINUATIONStudy drug was continued from the mid-trimester (20 + 0 wk and 23 + 6 wk of gestation) until 36 + 6 weeks of gestation, rupture of membranes, or delivery, whichever occurred first. Both the VP gel (Prochieve 8%, also known as Crinone 8%) and placebo were supplied (Columbia Laboratories, Inc.) as a soft, white to off-white gel, in a single-use, one-piece, white, disposable, polyethylene vaginal applicator with a twist-off top. The progesterone and placebo gels were identical in appearance. Each applicator delivered 1.125 g gel containing 90 mg progesterone or placebo and was wrapped and sealed in unmarked foil over-wrap. Both the active drug and the placebo were supplied in boxes of 14 applicators and were labeled with a unique kit number. Patients received a 2-week supply at randomization and at each subsequent visit.
MANAGEMENT OF AN EPISODE OF PRETERM LABORPatients who developed preterm labor during the study were treated according to the standard practice of the participating institutions, for example, admission to the hospital, bed rest, intravenous fluids, tocolytic therapy, and steroid administration, if clinically indicated. Administration of the study drug was to be continued during treatment for preterm labor until delivery (in the absence of preterm rupture of membranes). Maternal and neonatal outcomes were recorded throughout study participation and after delivery and discharge, using a standardized electronic reporting template.
EMERGENCY CERCLAGEAn emergency cerclage was allowed after randomization if the following criteria were met: (1) 21 to 26 weeks of gestation, (2) cervical dilation >2 cm, (3) membranes visible, (4) intact membranes, and (5) absence of uterine contractions, clinical chorioamnionitis, and significant vaginal bleeding.
PRIMARY AND SECONDARY OUTCOMESThe primary outcome of this study was preterm birth before 33 weeks of gestation. The key secondary outcome was neonatal morbidity, including respiratory distress syndrome (RDS), bronchopulmonary dysplasia, grade III or IV intraventricular hemorrhage, periventricular leukomalacia, proven sepsis, necrotizing enterocolitis, and perinatal mortality (fetal death or neonatal death). Four composite outcome scores were also used to assess perinatal mortality and neonatal morbidity (any event, two 0 to 4 scales, and one 0 to 6 scale). The definitions for individual outcomes and composite scores were prespecified. The outcome scores (0 to 4, 0 to 6) assigned ordinal values based upon the number of morbid events from 0 to 3 or 0 to 5; the highest number, 4 or 6, was assigned to a mortality event. For one of the 0 to 4 scores, the number of NICU days was also used for the assignment of the ordinal value. Other prespecified secondary outcomes included preterm birth <28, 35, and 37 weeks of gestation, neonatal length, weight, head circumference at birth, and incidence of congenital abnormalities. All outcomes were determined, and the database was locked before the unsealing of the randomization code.
SAMPLE SIZE CALCULATIONWe estimated that a sample size of 450 women (225 per treatment group) would have >90% power (2-tailed alpha level of 0.05) to detect a 55% reduction in the rate of preterm birth before 33 weeks of gestation, from 22% in the placebo group to 9.9% in the VP group.
STATISTICAL ANALYSESThe primary end point of the study, preterm birth before 33 weeks, was analyzed using the Cochran-Mantel-Haenszel test. The P value was assessed at the 2-sided significance level of 5%. Analysis of the primary efficacy end point was also performed using multivariable logistic regression, in which the following variables were included: treatment group, pooled study site, risk strata, gestational age at first dose, maternal age, cervical length, body mass index, and race.
Analysis of the trial was conducted in 3 population subsets:
Intent-to-treat analysis set: all patients were randomized to either VP gel or placebo; patients without a documented delivery date were excluded. Treated patient analysis set: patients who took at least one dose of either placebo or progesterone gel; women who received placebo and had no documented delivery date were considered as if they had delivered at term (37 wk of gestation); for women who received VP gel and had no documented delivery date, the date of last contact was used as the delivery date. Compliant analysis set: patients who used at least 80% of study medication, did not have a cerclage, and were not lost to follow-up. DATA AND SAFETY MONITORING BOARD AND TRIAL REGISTRATIONAn independent Data and Safety Monitoring Board reviewed unblinded data relevant to safety (not efficacy) after ~50% of the patients had been delivered. The observed frequency of adverse events did not exceed that expected or that stated in the informed consent. The Data and Safety Monitoring Board recommended the study continue without modification of the protocol or informed consent. This trial was registered with ClinicalTrials.gov, number NCT00615550.
INFORMED CONSENT AND HUMAN INVESTIGATIONS COMMITTEE APPROVALThe trial was reviewed and approved by the Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) of the National Institutes of Health (NIH) as the trial was sponsored by the Perinatology Research Branch (PRB) of NICHD/NIH. Approval was obtained from Wayne State University’s Human Investigation Committee as the main clinical site in the United States, as well as from each performing site. The approved informed consent was translated into the native languages of patients at the participating centers.
CLINICAL TRIAL AGREEMENTThe trial represented a partnership between NICHD and Columbia Laboratories, Inc. A clinical trial agreement was executed between the two entities.
MONITORING OF THE TRIALAn independent data coordinating center was responsible for randomization and data management. Clinical research monitors (Venn Life Sciences, St. Laurent, Quebec, Canada, and PharmOlam International, Houston, TX) conducted planned, regular site visits at each center, beginning with a site initiation visit and continuing until study completion, to independently assess compliance with the study protocol, timely collection of data, quality control, data completeness, and data accuracy, according to the International Conference on Harmonization and the U.S. Food and Drug Administration (FDA) Guidelines for Good Clinical Practice.93,94
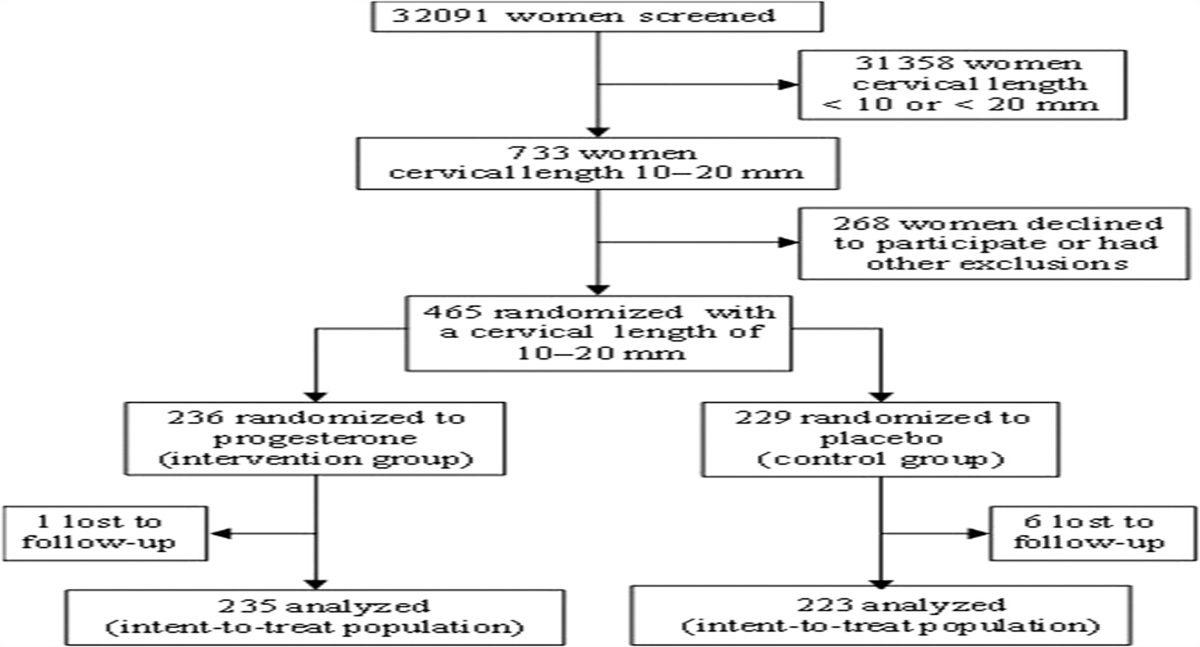
ENROLLMENT AND DISPOSITION OF PATIENTSThe trial was conducted between March 2008 and November 2010 and included 44 centers in 10 countries. Figure 1 illustrates the participant flow diagram. Of the 32,091 women who underwent sonographic measurement of cervical length between 19 + 0 and 23 + 6 weeks of gestation, 2.3% (733/32,091) were reported to have a cervical length of 10 to 20 mm.
 FIGURE 1: Participant flow diagram (reused with permission2).
FIGURE 1: Participant flow diagram (reused with permission2).Four hundred sixty-five women agreed to participate and were randomized, of whom 7 were lost to follow-up (VP gel, n = 1; placebo, n = 6). Thus, 458 women were included in the intent-to-treat analysis set (VP gel, n = 235; placebo, n = 223). Of the 458 women, 16% (n = 72) had a history of a previous preterm birth between 20 weeks and 35 weeks of gestation. Baseline maternal characteristics were similar between the placebo and the VP groups. Sixteen women (10 in the VP group and 6 in the placebo group; P = 0.46) underwent an emergency cervical cerclage after randomization.
RESULTS FOR THE PRIMARY END POINTPatients allocated to receive VP gel had a significantly lower rate of preterm birth before 33 weeks of gestation compared to those allocated to placebo [8.9% (21/235) vs 16.1% (36/223); RR, 0.55; 95% CI, 0.33-0.92; P = 0.02; adjusted (pooled study site and risk strata) RR, 0.54; 95% CI, 0.33-0.89; P = 0.01]. We estimated that 14 patients with a cervical length between 10 mm and 20 mm would need to be treated with VP gel to prevent one case of preterm birth before 33 weeks of gestation (95% CI: 8-87).
The primary end point was also significant after adjustment for pooled study site, risk strata, treatment group, gestational age at first dose, maternal age, cervical length, body mass index, and race using multivariable logistic regression analysis (adjusted RR, 0.52; 95% CI, 0.31-0.91; P = 0.02). No interaction between the treatment and pooled study site was detected (P = 0.2). In women without a history of preterm birth (84% of the population), VP gel administration was associated with a significant reduction in the rate of preterm birth before 33 weeks [7.6% (15/197) vs 15.3% (29/189); RR, 0.50; 95% CI, 0.27-0.90; P = 0.02 (Table 1)]. VP gel was also associated with a significant reduction in the rate of preterm birth before 35 weeks [14.5% (34/235) vs 23.3% (52/223); RR, 0.62; 95% CI, 0.42-0.92; P = 0.02] and before 28 weeks of gestation [5.1% (12/235) vs 10.3% (23/223); RR, 0.50; 95% CI, 0.25-0.97; P = 0.04].
TABLE 1 - Gestational Age at Delivery and Neonatal Outcome in Asymptomatic Women With a Singleton Pregnancy and Sonographic Short Cervix Allocated to Receive VP Gel (n = 235) Compared to Those Allocated to Receive Placebo (n = 223): Intent-to-Treat Analysis Set VP Placebo Relative risk Outcome n (%) n (%) 95% CI P Primary outcome Preterm birth <33 wk 21/235 (8.9) 36/223 (16.1) 0.55 (0.33–0.92) 0.020 Secondary outcomes Preterm birth <28 wk 12/235 (5.1) 23/223 (10.3) 0.50 (0.25–0.97) 0.036 Preterm birth <35 wk 34/235 (14.5) 52/223 (23.3) 0.62 (0.42–0.92) 0.016 Preterm birth <37 wk 71/235 (30.2) 76/223 (34.1) 0.89 (0.68–1.16) 0.376 RDS 7/235 (3.0) 17/223 (7.6) 0.39 (0.17–0.92) 0.026 Bronchopulmonary dysplasia 4/235 (1.7) 5/223 (2.2) 0.76 (0.21–2.79) 0.678 Proven sepsis 7/235 (3.0) 6/223 (2.7) 1.11 (0.38–3.24) 0.853 Necrotizing enterocolitis 5/235 (2.1) 4/223 (1.8) 1.19 (0.32–4.36) 0.797 Intraventricular hemorrhage, grade III/IV 0/235 (0.0) 1/223 (0.5) 0.32 (0.01–7.73) 0.305 Periventricular leukomalacia 0/235 (0.0) 0/223 (0.0) Not estimable NA Perinatal death 8/235 (3.4) 11/223 (4.9) 0.69 (0.28–1.68) 0.413 Fetal death 5/235 (2.1) 6/223 (2.7) 0.79 (0.25–2.57) 0.700 Neonatal death 3/235 (1.3) 5/223 (2.2) 0.57 (0.14–2.35) 0.431 Composite outcome scores Any morbidity/mortality event 18/235 (7.7) 30/223 (13.5) 0.57 (0.33–0.99) 0.043 Birthweight <2500 g 60/234 (25.6) 68/220 (30.9) 0.83 (0.62–1.11) 0.213 Birthweight <1500 g 15/234 (6.4) 30/220 (13.6) 0.47 (0.26–0.85) 0.010Unadjusted relative risk and 95%CI were calculated using the Cochran-Mantel-Haenszel test.
NA indicates not available; RDS, respiratory distress syndrome; VP, vaginal progesterone.
Figure 2 displays the survival analysis for patients in the entire intent-to-treat analysis set (Fig. 2A), patients with no prior preterm delivery (Fig. 2B), and patients with a prior preterm delivery (Fig. 2C). The curves demonstrate a separation between patients allocated to receive VP gel and those in the placebo group. Importantly, there was no difference in the proportion of patients who delivered <37 weeks of gestation because the curves converge and overlap at this point. One interpretation of this finding is that the administration of VP shifted the proportion of patients who would have delivered very preterm to a later gestational age.
 FIGURE 2: Survival analysis of intent-to-treat analysis set showing the proportion of patients remaining undelivered according to treatment allocation: vaginal progesterone (VP; ——) versus placebo (- - - - ). A, Entire population (patients with and without a prior history of preterm delivery; VP, n = 235; placebo, n = 223). B, Patients without a prior history of preterm delivery (VP, n = 197; placebo, n = 189). C, Patients with a prior history of preterm delivery (VP, n = 38; placebo, n = 34). P >0.05 for all comparisons (reused with permission2).NEONATAL OUTCOMES
FIGURE 2: Survival analysis of intent-to-treat analysis set showing the proportion of patients remaining undelivered according to treatment allocation: vaginal progesterone (VP; ——) versus placebo (- - - - ). A, Entire population (patients with and without a prior history of preterm delivery; VP, n = 235; placebo, n = 223). B, Patients without a prior history of preterm delivery (VP, n = 197; placebo, n = 189). C, Patients with a prior history of preterm delivery (VP, n = 38; placebo, n = 34). P >0.05 for all comparisons (reused with permission2).NEONATAL OUTCOMES
Neonates born to women allocated to receive VP gel had a significantly lower frequency of RDS than did those born to women allocated to receive placebo [3.0% (7/235) vs 7.6% (17/223); RR, 0.39; 95% CI, 0.17-0.92; P = 0.03]. The number needed to treat for benefit was 22 (95% CI: 12-186). This effect remained significant after adjustment for pooled study sites and risk strata (RR, 0.40; 95% CI, 0.17-0.94; P = 0.03). VP was associated with a significant reduction in the rate of neonatal birthweight <1500 g [6.4% (15/234) vs 13.6% (30/220); RR: 0.47; 95% CI: 0.26-0.85; P = 0.01].
The rate of the composite outcome of any morbidity or mortality was significantly lower in the neonates born to mothers allocated to receive VP gel compared to those born to mothers allocated to receive placebo [7.7% (18/235) vs 13.5% (30/223); RR, 0.57; 95% CI, 0.33-0.99; P = 0.04]. The composite scores “0 to 4 scale without NICU” and “0 to 6 scale without NICU” were also significantly lower in the progesterone gel group compared to the placebo group (P < 0.05 for both comparisons). After adjustment for pooled study site and risk strata, the effect of VP gel on composite perinatal mortality/neonatal morbidity scores “any morbidity/mortality event,” “0 to 4 scale without NICU,” and “0 to 6 scale without NICU” continued to show trends toward improvement (P = 0.054, 0.065, and 0.065, respectively).
Adverse EventsThe frequency of adverse events was comparable between patients who received VP gel and those who received placebo. The rate of adverse events related to study treatment was not significantly different in women who received VP gel compared to those who received placebo [12.8% (30/235) vs 10.7% (24/224); RR, 1.14; 95% CI, 0.72-1.80; P = 0.59]; the most frequently reported adverse events related to study treatment occurred in up to 2% of women and included vaginal pruritus, vaginal discharge, vaginal candidiasis, and nausea. Furthermore, no fetal or neonatal safety signal was detected for VP gel.
Sensitivity Analysis Based on the “Treated Patient Analysis Set”The first sensitivity analysis was performed in patients who had received at least one dose of either placebo or progesterone. VP decreased the rate of preterm birth before 33 weeks of gestation after adjustment for study site and risk strata [8.9% (21/235) vs 15.2% (34/224); RR, 0.56; 95% CI, 0.33–0.93; P = 0.02], as well as the rate of RDS [3.0% (7/235) vs 7.1% (16/224); RR, 0.42; 95% CI, 0.18–0.97; P = 0.04].
Sensitivity Analysis Based on the “Compliance Data Set”A prespecified analysis was conducted in the subgroup of patients of the treated patient analysis set, excluding those who had <80% treatment compliance (n = 53), those who did not have a documented delivery date (n = 4), or those who had a cerclage (n = 17). This compliance analysis set showed that patients allocated to VP gel had a significantly lower frequency of preterm birth than did those allocated to placebo for delivery <28 weeks of gestation [3.1% (6/194) vs 7.8% (15/193); RR, 0.40; 95% CI, 0.16–1.00; P = 0.04], delivery <33 weeks of gestation [5.7% (11/194) vs 13.0% (25/193); RR, 0.44; 95% CI, 0.22–0.86; P = 0.01] and delivery <35 weeks of gestation [10.3% (20/194) vs 20.2% (39/193); RR, 0.51; 95% CI, 0.31–0.84; P < 0.01]. There was no significant difference in the rate of preterm delivery before 37 weeks of gestation [26.8% (52/194) vs 30.6% (59/193); RR, 0.88; 95% CI, 0.64–1.20; P = 0.41]. After adjustment for study site and risk strata, the effect of VP gel remained significant for the reduction in the primary end point—the rate of preterm birth <33 weeks of gestation (RR, 0.42; 95% CI, 0.22–0.82; P < 0.01) and preterm birth <35 weeks of gestation (RR, 0.50; 95% CI, 0.31–0.82; P < 0.01).
Noteworthy Features of the PREGNANT TrialTwo features of the PREGNANT trial make it different from other studies: (1) the primary end point was delivery <33 weeks of gestation and (2) the criteria for cervical length restricted inclusion to patients with a cervical length of 10 to 20 mm.
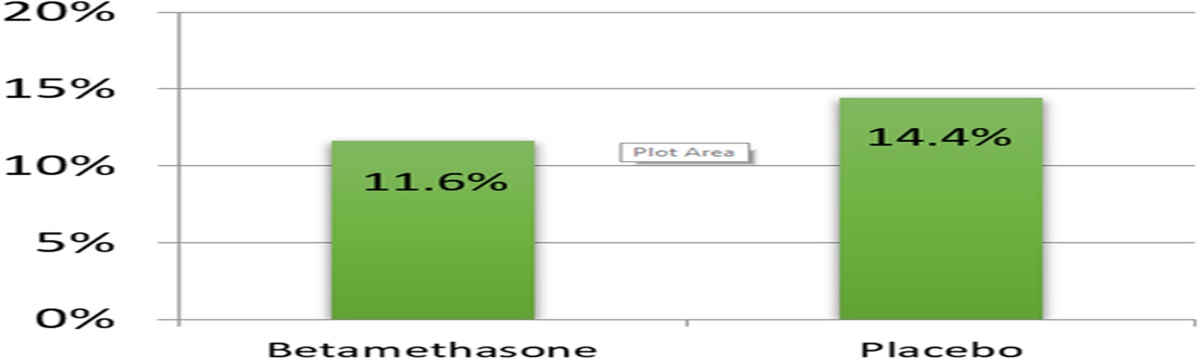
The rationale for using delivery <33 weeks for the primary end point rather than the conventional definition of preterm birth <37 weeks was a crucial one. If we had used preterm delivery <37 weeks, the trial would have been negative (Fig. 3). The PREGNANT trial and subsequent metanalyses showed that VP prevents earlier preterm delivery better than late preterm delivery. Indeed, there was a 45% reduction in preterm delivery <33 weeks (P = 0.02), a 50% reduction in preterm birth <28 weeks (P = 0.036), but only a 38% reduction in preterm birth <35 weeks of gestation. Importantly, there was no reduction in the rate of preterm birth if the outcome measure is birth <37 weeks of gestation. Prevention of earlier preterm birth is desirable because of the high rate of morbidity and mortality associated with early preterm birth. Preterm parturition is a syndrome caused by multiple mechanisms of disease, and it is not surprising that an intervention such as VP will prevent some, but not all, forms of spontaneous preterm birth. The most common pathologic feature in late preterm birth is chronic chorioamnionitis,95 a lesion that is the histopathologic hallmark of maternal anti-fetal rejection.95,96 It is possible that VP is not effective in this subgroup of patients, but, instead, it prevents the progressive cervical shortening that leads to early preterm birth.
 FIGURE 3:
FIGURE 3: The rate of preterm delivery at different gestational ages: PREGNANT trial.
The selection of preterm delivery <33 weeks of gestation as the primary end point was also based on another study design consideration. A prior study by Fonseca et al,97 which included 250 women and used spontaneous preterm delivery <34 weeks as a primary end point, found a significant reduction in this outcome measure but no significant differences in preterm delivery <28 weeks and <37 weeks of gestation and in neonatal morbidity despite a clear trend toward reduction. Therefore, critics argued that the reduction in preterm birth did not result in a beneficial effect for neonates, which is the key goal in reducing the rate of preterm birth. We chose preterm delivery <33 weeks as a primary end point, which required a large sample size (about 450), and this increased the probability and the detection of differences in RDS, the most common neonatal complication, as well as for other secondary end points.
Another noteworthy feature of the PREGNANT trial was the criterion of a sonographic short cervix. The tenth percentile for cervical length in the mid-trimester is 25 mm.98 However, instead of choosing a single cutoff value (eg, <15 mm, <25 mm, or <30 mm), we elected to restrict the cervical length to 10 to 20 mm. The rationale for this was to improve the efficiency of the trial by including patients most likely to benefit from VP. A previous trial of VP in women at cervical length <15 mm had shown a significant reduction in the rate of preterm birth in patients allocated to VP with an overall reduction of 49%. However, the efficacy was different according to cervical length. Among women with a cervical length of 1 to 5 mm, VP reduced the rate of preterm birth by only 15%, whereas in women with a cervical length of 6 to 10 mm, the effect size was 25%. By contrast, in patients with a cervical length of 11 to 15 mm, there was a 75% reduction in the rate of preterm birth. This prior knowledge and the information available about the pathophysiology of preterm birth were instrumental in formulating the criteria for inclusion. For example, we observed that the frequency of microbial invasion of the amniotic cavity (defined as a positive amniotic fluid culture) was 9% in patients with an asymptomatic short cervix.99 We also observed that in patients with a nonmeasurable cervix <24 weeks of gestation, the rate of intra-amniotic inflammation (defined as an elevated IL-6 level in amniotic fluid) was high (19%), and pregnancy outcome was poor. Therefore, we reasoned it may be best to exclude patients with an extremely short cervix (<10 mm) in whom VP would be less likely to be effective as there is no evidence that progesterone can treat intra-amniotic infection or intra-amniotic inflammation.
We also excluded patients who had a cervical length >20 mm. The rate of spontaneous preterm delivery varies with cervical length and the longer the cervix, the lower the rate of preterm delivery.100–102 Excluding patients of >20 mm was intended to improve trial efficiency and reduce cost. This, of course, does not mean progesterone was not effective in patients with a longer cervical length (20 to 25 mm). At the time, we considered that this question could be addressed subsequently if the PREGNANT trial was positive.
A Public/Private PartnershipThe PREGNANT trial was a partnership between the PRB of the NICHD and Columbia Laboratories, Inc. The PRB had been interested in the etiology of spontaneous preterm birth, prediction, and prevention. Columbia Laboratories, Inc. had a preparation of VP that had been approved for other indications by the FDA. Columbia Laboratories, Inc. sponsored studies to explore the value of VP in the prevention of preterm birth in patients with a history of spontaneous preterm birth or those with an increased risk.
The Food and Drug Administration Review of the PREGNANT TrialThe results of the PREGNANT trial were submitted by Columbia Laboratories to the FDA for a New Drug Application. The purpose of the application was to obtain approval to market the drug in the United States. The FDA staff reviewed the application and presented an analysis to a group of experts called an Advisory Group. The initial statistical analysis presented by the statisticians of the FDA concluded that the data “did not support the efficacy of progesterone gel in the prevention of preterm birth.”103 The authors of the PREGNANT trial reported to the FDA that VP reduced the risk of preterm delivery, using the statistical analysis plan that had been agreed upon before the unmasking of the trial. The relative risk was 0.56 (95% CI, 0.33-0.93; P = 0.022). By contrast, the FDA did not use a prespecified analysis plan and did a post hoc analysis adjusting for the geographical region (U.S. vs non-U.S., maternal age, and cervical length). This post hoc analysis is neither the standard approach to analyze randomized trials nor is it appropriate. A body of literature is available demonstrating the inappropriateness of this approach as illustrated by Yusuf and Wittes.104 The authors list examples of large differences in trial results according to geographic regions. They indicated that “geography does not trump biology in this case,” and they would have applied the overall results of the trial to the United States.
It is valuable for practitioners of obstetrics to be aware of the pitfalls in the analysis of randomized trials when post hoc analyses by geographic region are performed. We will illustrate this with an example and by using one of the best-known cases. An important randomized trial tested the effects of intravenous streptokinase and oral aspirin, both or neither, in patients with a suspected acute myocardial infarction (Second International Study of Infarct Survival).105
留言 (0)