
記住我






Our report follows Standards for Reporting Diagnostic Accuracy (STARD) guidelines for reporting diagnostic studies [8] (Online supplement 1).
Study design and settingDuring screening for eligibility in a pilot randomised controlled trial (RCT) (ANZCTR number ANZCTRN12619001530112) which commenced in September 2019, pregnant women were invited to undertake level III Apnealink Air in the home environment, and level I attended PSG in a hospital sleep investigation unit (SIU) by the 24th week of gestation. SARS-CoV-2 pandemic public health orders in South-Western Sydney during 2020 resulted in the closure of the SIU and temporary suspension of the study and we were unable to complete attended PSG during this time. During the study recruitment pause we developed an amended protocol to include validation of an additional sleep diagnostic test (Somte). Somte were to be completed in the home environment using self-application method to reduce staff exposure to patients, which was the public mandate at the time. Data were to be compared to gold-standard attended PSG. The amendment was approved in January 2021, implemented in March 2021 and the first Somte study collected 28th March 2021. A further SARS-CoV-2 study suspension and SIU closure restricted access to attended PSG between July and December 2021. Sample size target for this study was not predetermined. The intention was to collect as many paired studies as possible during the study collection period. Recruitment for this study concluded in January 2023.
Participants were recruited from antenatal clinics at Campbelltown and Liverpool hospitals in South Western Sydney, NSW, Australia. Somte tests were completed in the participant home. Attended PSG were completed in the SIU at Liverpool Hospital. Participants diagnosed with OSA on PSG were offered participation in the RCT study if eligible. Participants not eligible for RCT participation but who were deemed by the sleep physician to require treatment were offered CPAP through a clinical pathway.
ParticipantsAt or prior to scheduled hospital obstetric bookings, participants were approached in person or by phone in a convenience series by study staff who completed screening eligibility using REDCap [9]. Eligible participants were provided a participant information sheet by email or hard copy and provided informed written consent if eligible to participate. Baseline data were collected using REDCap questionnaire, including STOP BANG [10], Epworth Sleepiness Scale (ESS) [11] and pregnancy-related tool questionnaires [12]. Gestation was based on the dating scan performed in the first trimester. Participants undertook both the Somte and PSG according to availability of participant and test devices which were organised by the study coordinator. Both studies were scheduled within a target period of 7 days. Only participants who successfully completed both tests within 14 days were included for analysis.
Inclusion criteria were defined as (1) women aged 18 years of age and above; (2) In early-mid pregnancy (up 24 weeks gestation); (3) at increased risk of metabolic complications defined as ONE OR MORE of: (a) body mass index (BMI) greater than or equal to 35 kg/m2 at screening; (b) previous Gestational diabetes mellitus (GDM) [13]; (c) previous personal history of pre-eclampsia (or in mother or sister); (d) underlying renal disease; (e) maternal type 2 diabetes (pre-gestational); (f) symptoms of sleep disordered breathing (SDB) including snoring, witnessed apnoea, mild excessive daytime sleepiness (EDS) (which does not meet the criteria for severe EDS defined by Epworth Sleepiness Scale (ESS) (> 15) or a fall asleep accident or near-miss accident in the previous 12 months) or tiredness. Participants were excluded if they have (1) previous diagnosis of OSA on active treatment; (2) confirmed current GDM or preeclampsia; (3) maternal type 1 diabetes; (4) multifetal gestation, (5) known fetal chromosomal abnormality; (6) inability to provide informed consent; (7) severe EDS based on clinical assessment (e.g. including a fall asleep motor vehicle accident or near miss, transient sleepiness while driving/at lights or needing to pull over due to sleepiness while driving, or transient sleepiness in any other dangerous situation i.e. cooking, carrying baby) or ESS of greater than 15.
Attended PSG (reference standard)Participants completed PSG (Grael 4 K PSG: EEG, Grael Acquisition system, Compumedics, Abbotsford, Australia). Participants had routine observations on arrival (BP, pulse, Sp02, height, weight) and if there were any clinically significant abnormalities, an obstetric physician was called. Participants were set up by experienced sleep technicians. PSG data were collected between 21:00–06:00 approximately. PSG data collection followed international 10–20 system and included EEG (C1/C2 C3/C4, O1/O2, F3/F4, M1/M2), EOG (E1/E2), EMG (chin, diaphragm and anterior tibialis (left and right)), snore (microphone), ECG (modified Lead II), airflow (pressure transducer, thermistor), respiratory effort (abdomen and thoracic), oximeter (SpO2), sound level (dB meter), digital video (audio and visual) and position sensor (Fig. 1b). SpO2 was collected using Compumedics adult silicone soft tip probe- oximeter- 3 m. Time available for sleep (TAS) represents recording time in minutes between lights on and off / off and on, as visualised in study video. Total sleep time (TST) represents sleep time in minutes.
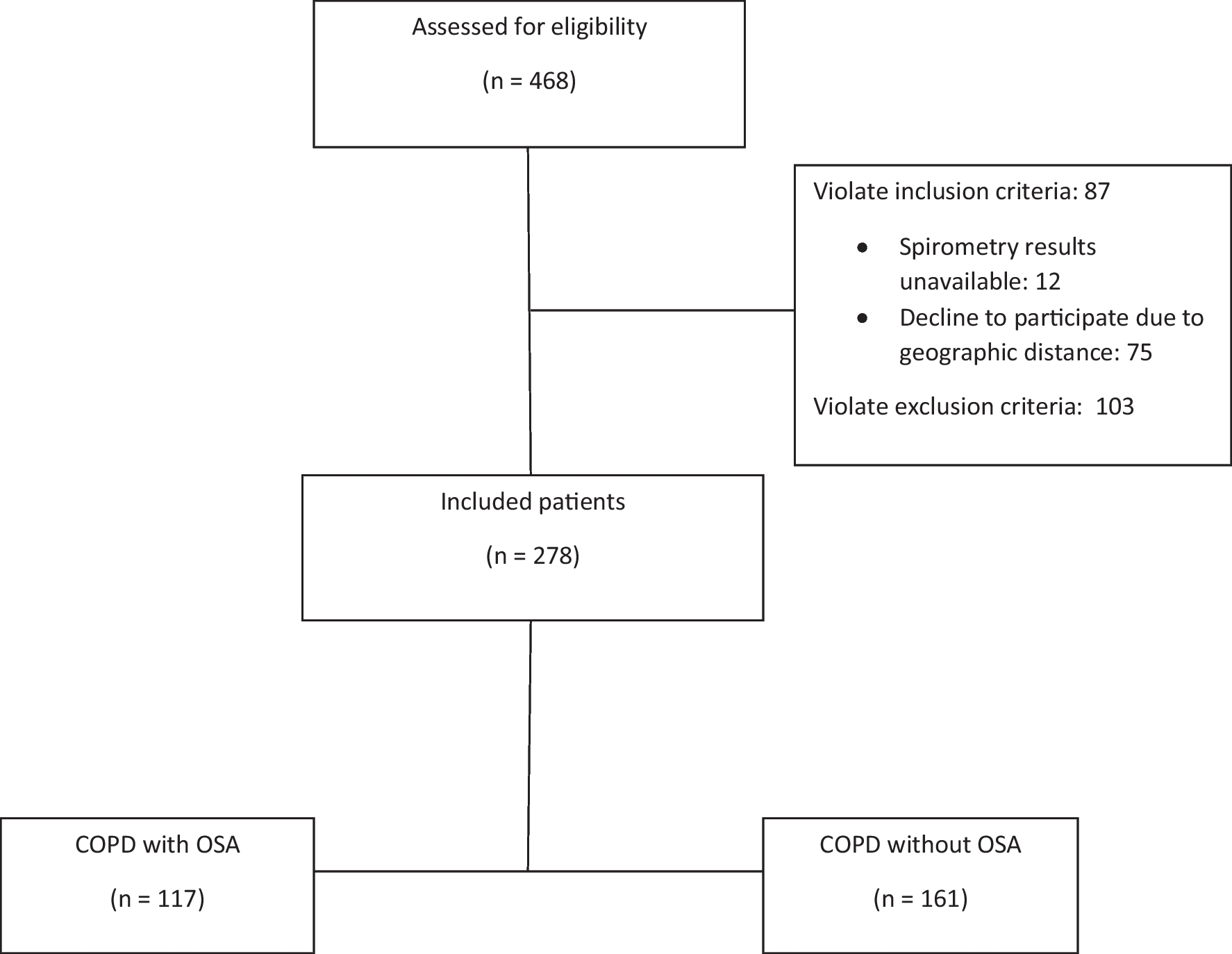
Fig. 1
a, b Devices used in the study. Figure 1a, b demonstrates the first author wearing the Somte and PSG devices. The Somte image does not depict the thermistor, which was used during the test. The Somte was completed in the participant home, following self-application of the test. The PSG was completed in the sleep investigations unit at Liverpool Hospital, following technician application of the test
Somte (index test)Participants were issued the Somte (Compumedics, Abbotsford, Australia) and manufacturer instructional booklet by the study coordinator at the consent visit if the test was scheduled to be completed on the night of the consent visit, or on the date of the Somte study, if it was scheduled to be completed later. Participants were encouraged to watch a manufacturer produced YouTube instructional video prior to undertaking the test [14]. The study coordinator demonstrated the Somte device operation and correct application technique for the nasal cannula, thermistor, oximeter, and abdominal and thoracic effort bands. Participants self-applied the Somte device in the home environment. Assistance from spouse or partner, during the set-up of Somte, was allowed, but not encouraged.
Somte data collection commenced using either manual or pre-specified start options. Initially, participants manually started the Somte at their preferred sleep time, using the manual start function on the device. However, due to failure to start in four Somte studies, start times were subsequently preprogramed according to the anticipated sleep time, as reported by the participant on the day of device collection. The study coordinator was available for technical phone support during the sleep study set-up and data collection. Participants were instructed to manually stop the test at end of sleep period (sleep offset).
Somte data collection followed a modified American Academy of Sleep Medicine (AASM) set-up protocol. Participants self-applied EEG (F3/F4, M1/M2), electrooculography (EOG (E1/E2)), EMG chin (submentalis, anterior tibialis (left and right leg), electrocardiogram (ECG (modified Lead II)), airflow (pressure transducer), snore, airflow (thermistor), respiratory effort (abdomen and thoracic), oximeter (SpO2) and position sensor. F3/4 leads were placed on the forehead for ease of application by the participant (Fig. 1a). SpO2 was collected using Compumedics adult silicone soft tip probe- oximeter- 1 m. Time available for sleep (TAS) represents recording time in minutes of the duration of recording between which the scorer deemed the patient to have attempted to sleep, and was determined using a combination of position change, signal quality and movement. Our study did not collect patient reporting of sleep onset and sleep offset time via questionnaire. Total sleep time (TST) represents sleep time in minutes.
Scoring and reporting of studiesSomte and PSG were manually scored using version 2.6 2020 AASM manual for the scoring of sleep and associated events [15] using Profusion PSG 4 software (Compumedics, Abbotsford, Australia). Event definitions are defined according to AASM guidelines:
Obstructive apnoea: reduction of ≥ 90% of baseline in airflow for 10 s or more with continued or increased effort throughout.
Central apnoea: reduction of ≥ 90% of baseline in airflow for 10 s or more with absent effort throughout.
Mixed apnoea: reduction of ≥ 90% of baseline in airflow for 10 s or more with absent effort initially followed by resumption of effort in the second portion.
Hypopnoea: reduction of ≥ 30% of baseline in airflow for 10 s or more accompanied by either
an arousal or 3% desaturation.
Respiratory Effort-Related Arousal (RERA): a sequence of breaths lasting ≥ 10 s characterized by increasing effort or by flattening of the inspiratory portion of the flow leading to an arousal from sleep.
AHI: Apnoeas and hypopnoeas only.
Respiratory Disturbance Index (RDI): AHI + RERA.
Somte were de-identified prior to data collection. Following data collection, Somte raw data were downloaded from the device by the study coordinator and scored and reported blinded to the PSG result. A single, experienced sleep scientist manually scored all Somte and PSG. Following scoring, one of two sleep physicians who were not involved in the direct care of respective patients, reported studies. The study coordinator was unblinded to identified participant sleep data though was not involved in the scoring or reporting of any Somte or PSG studies.
Statistical methodsData were entered into REDCap. Analysis was performed using Statistical Package for the Social Sciences (SPSS), Version 29.0 (IBM Corp., Chicago, Illinois, USA). Data were checked for errors in SPSS by calculating minimum and maximum values for each variable and checking for outliers or anomalies. Descriptive data is presented (based on distribution as assessed by a Shapiro–Wilk test) either as a mean ± standard deviation (SD) or median (interquartile range (IQR)) or count (%). Diagnostic test accuracy measures were undertaken (sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV)) and by creating receiver operating characteristic (ROC) curves. Time available for sleep (TAS) and total sleep time (TST) of both tests were calculated and reported in minutes. Cohen's kappa coefficient was calculated. Sleep variable data of both tests were calculated and reported in percentage of TST, events per hour, minimum or mean. The calculated AHI and RDI of each method were compared and Bland–Altman plot used to plot limits of agreement. The AHI was the primary outcome of interest though an AHI ≥ 5 or RDI ≥ 5 at PSG was considered diagnostic of OSA for the purpose of eligibility screening for participation in the RCT. Imputation was not used for missing data. Indeterminate results were considered false-positive or false-negative and incorporated into the final analysis.
留言 (0)