

記住我




The current list of inborn errors of immunity (IEI) comprises more than 485 monogenetic gene defects (1). Enhanced susceptibility to a specific pathogen such as Staphylococcus aureus (S. aureus) may raise suspicion of a certain type of immunological impairment. Staphylococcus aureus is a great challenge to our health care systems (2). Despite being considered a commensal, with a colonization rate of 20%–30% in the healthy population (3), it can also cause a wide variety of different infections. It is a leading cause of skin and soft tissue infections and abscesses, but may also lead to lung infections, osteomyelitis or endocarditis, in particular in patients with underlying conditions (2). The ability to colonize but also to cause harm to the host, emerges from a complex interaction between the pathogen and its host (4). Staphylococcus aureus is a specialist in adapting to the human host by evading almost every aspect of the immune system (5). In the last decades, changes in strains have led to an increase of S. aureus infections in otherwise healthy individuals (6). Thus, staphylococcal defense in the individual is shaped by both pathogen virulence factors as well as the patient's immune predisposition (4). Recurrent or severe S. aureus infections may both be an indicator of certain IEI and specific IEI can teach us about essential immune functions for staphylococcal defense.
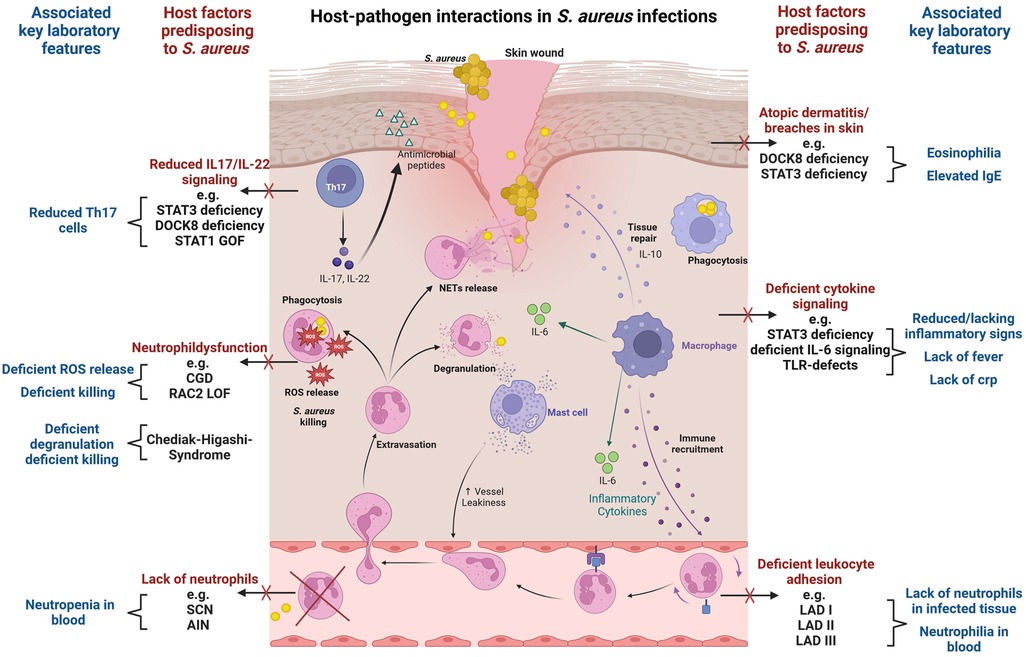
S. aureus immune evasion and host immune responseStaphylococcal infections often arise from asymptomatic colonization and breaches through skin and mucosal barriers (7) (Figure 1). Immune evasion strategies of S. aureus are abundant and tackle particularly innate immunity (8, 9). Examples include inhibition of immune recognition, prevention of complement activation (10), resistance to phagosomal killing (5) and direct killing of immune cells through different leucocidins (7). In addition, presence of peptidoglycan layer, polysaccharide capsule and surface proteins hamper opsonization (7). The most important players in S. aureus defense are phagocytes. In particular neutrophils, along with tissue-resident or monocyte-derived macrophages, are instrumental in identifying, engulfing, and eliminating staphylococci (11). As the first line of innate cellular defense, they also orchestrate subsequent immune responses. The crucial role of neutrophils is clearly evidenced by the enhanced staphylococcal susceptibility of patients with numeric or functional neutrophil defects (12, 13). Staphylococcus aureus has developed numerous mechanisms to reduce neutrophil extravasation, activation, and chemotaxis (9), and may also evade neutrophil extracellular traps using nucleases and proteases (14). Secretion of exopolysaccharides and biofilm formation inhibit phagocytosis (7). When internalized by phagocytes, S. aureus may neutralize reactive oxygen species and employ enzymes for survival (8). Through intracellular survival both in phagocytic and non-phagocytic cells, S. aureus may evade antibiotic killing and facilitate subsequent dissemination (15). Induction of IL-10 by S. aureus may lead to a phenotypic switch in the immune response during persistent staphylococcal infection allowing its persistence as commensal (16). Toxins like Panton–Valentine leucocidin (PVL), which are harbored by some more virulent strains, destroy immune cells and may lead to treatment failure and severe infections even in immunocompetent patients (17, 18). While most virulence factors address innate immunity, S. aureus may also interfere with the adaptive immune response, using proteins like SpA to bind immunoglobulins (19) and superantigens like TSST-1 to induce cytokine release and toxic shock syndrome (20).
Figure 1. Host-pathogen interactions in S. aureus infections. The figure visualizes key immunological defense mechanisms and highlights host factors predisposing to S. aureus infection in case of deficiency. Commonly associated laboratory findings in the respective setting are also displayed. Selective examples of IEI with susceptibility to S. aureus infection are provided. The figure provides a simplified overview, and displayed host factors and interactions do not claim to be complete. The figure was created with BioRender.com.
The evasion strategies of S. aureus challenge infection management, prevention and vaccine development (8). We provide an overview of IEI that render individuals susceptible to S. aureus infections (Table 1 and Supplementary Table S1), highlighting key immunological defense mechanism involved in staphylococcal immunity.
Table 1. IEI with recurrent or severe S. aureus infections.
IEI with low neutrophil numbers and susceptibility to S. aureus infectionsSevere congenital neutropenia (SCN) is usually characterized by severe neutropenia (<500/µl) due to myeloid maturation arrest in the bone marrow. Over 20 different genes have been identified (21). Lack of mature neutrophils leads to a severe infectious phenotype with potentially life-threatening disease in the first months of life. Infections are caused not only by S. aureus but also by gram negative bacteria, and blood stream infections are common. Depending on the underlying gene defect there may be additional somatic features (Supplementary Table S1) (22).
Primary autoimmune neutropenia (AIN) of infancy, which is the most common type of neutropenia in childhood and may also present with nearly absent neutrophils and susceptibility to staphylococcal skin infections (abscesses, furunculosis), needs to be separated from SCN. AIN is typically detected in infancy, frequently as an incidental finding, and shows spontaneous remission in early childhood (23). Neutrophils mature normally in the bone marrow but peripheral numbers may be very low due to the presence of anti-neutrophilic antibodies. Infections are less severe compared to SCN. While the detection of anti-neutrophilic antibodies is suggestive of AIN it does not fully exclude additional SCN. Thus, in cases with severe infections or persistent neutropenia bone marrow evaluation and genetic testing may be indicated. If detected in older children or adults, AIN is more likely to be an immune phenomenon related to another IEI/autoimmune disorders requiring further diagnostic workup (24).
IEI with neutrophil function defects and susceptibility to S. aureus infectionsChronic granulomatous disease (CGD) represents the most common hereditary phagocyte dysfunction with an estimated prevalence of around 1:200,000 (25, 26). CGD leads to deficient reactive oxygen species (ROS) generation due to loss-of-function mutations affecting different aspects of the multicomponent enzyme NADPH oxidase in phagocytes (Nox2) (27). CGD patients experience severe infections accompanied by granuloma and abscess formation. Staphylococcus aureus is the most common pathogen isolated from skin infections/abscesses, liver abscesses and lymphadenitis, but it may also lead to pulmonary infections or sepsis. Patients are also very susceptible to Aspergillus spp. (26). Other characteristic pathogens in CGD include gram negative bacteria (e.g., Salmonella) and catalase positive bacteria (e.g., Burkholderia, Serratia and Nocardia) (12, 28). Additionally, CGD is associated with inflammatory complications like colitis, which might be related to defective T-cell regulation but also hyperactivation of NF-kB and inflammasome pathways (27, 29).
Leukocyte adhesion deficiency (LAD) is characterized by functional defects in neutrophil adhesion, integrin activation or rolling, leading to an inability to migrate effectively to infection sites (30). This results in a striking discrepancy with lack of pus formation at infection sites despite significant leukocytosis with neutrophilia in the blood. LAD patients typically experience recurrent bacterial and fungal infections, delayed wound healing, and other associated features (31). Three different genetic defects affecting neutrophils are known. Associated features are omphalitis and gingivitis (LAD I), developmental impairment and short statue (LAD II), and bleeding tendency (LAD III) (30, 32).
Combined IEI which frequently cause neutropenia or neutrophil dysfunctionNeutropenia has also been described in certain combined immunodeficiencies. Typical examples are CD40Ligand (CD40l) and CD40 deficiency, which are characterized by abnormal serum immunoglobulin levels due to impaired interaction between CD40l on T cells and CD40 on antigen-presenting cells (33, 34). These conditions lead to both impaired cellular and humoral immunity, which results in a broad infection phenotype. Patients frequently present with opportunistic infections (e.g., pneumocystis jirovecii, cryptosporidium, aspergillus spp.) (35). IgM may be elevated concomitantly to low IgA and IgG, which lead to bacterial respiratory and gastrointestinal infections (33). Intermittent or permanent neutropenia might be related to deficient release of growth factors important for granulopoiesis due to impaired CD40-CD40l-interaction (36). Furthermore, functional defects in neutrophils have been described in CD40l deficiency (37).
Mutations in Ras-related C3 botulinum toxin substrate 2 (RAC2) are also typically affecting neutrophil function. RAC2 is an essential regulator of neutrophil chemotaxis and contributes to NADPH oxidase function (38). Autosomal-dominant (AD) RAC2 loss of function (LOF) mutations cause LAD-like disease with neutrophilia and functional neutrophil defects (e.g., deficient chemotaxis and ROS generation) (39). In contrast, AD RAC2 gain of function (GOF) mutations lead to (severe) combined immunodeficiencies with lymphopenia and low immunoglobulins, frequent neutropenia and functional neutrophil abnormalities (38, 40).
Neutropenia has also been reported in some patients with deficiency in phosphoglucomutase 3 (PGM3), a disorder of glycosylation which is currently classified as autosomal-recessive Hyper IgE syndrome (1). PGM3 deficiency presents with eczema, eosinophilia, elevated IgE, but may also display a CID/SCID phenotype, facial dysmorphism and neurocognitive impairment (41).
Patients with autosomal-recessive deficiency of dedicator of cytokines (DOCK) 8 display severe atopic dermatitis with S. aureus colonization and skin infections (DOCK8 deficiency). Osteomyelitis has also been reported (42). DOCK8 plays a crucial role in lymphocyte proliferation, migration of dendritic cells, and generation of long-term memory in B- and T cells, thus predisposing patients to a mostly severe phenotype regarding viral and mycobacterial infections (43). Dysfunction of regulatory T-cells together with S. aureus exposure have been suggested to drive severe eczema in DOCK8 deficiency (44) and DOCK8-deficient murine neutrophils were prone to undergo S. aureus-induced cell death (45). In addition, reduced signal transducer and activator of transcription 3 (STAT3) signaling and low T helper 17 (Th17) cells have also been reported (46).
IEI with staphylococcal susceptibility associated to defective cytokine signalingAutosomal-dominant Hyper-IgE syndrome due to dominant-negative mutations in STAT3 (STAT3-HIES) is one of the key IEI associated with a specific susceptibility to S. aureus infections, particularly in the skin and lung (47). Recurrent “cold” abscesses with lacking systemic signs of infections are typical. STAT3 functions as a transcription factor downstream of the tyrosine kinases janus activated kinase (JAK)1, JAK2, and tyrosine kinase 2 (TYK2) and enables signal transduction through various cytokines, such as interleukin-6 (IL-6), IL-10, IL-11, IL-21, and IL-23 (48). STAT3 deficiency results in failure of Th17 cell differentiation (49). Th17 function has been shown to be pivotal in Candida defense (50), explaining the patients' predisposition to mucocutaneous candidiasis. Th17 cells aid epithelial cells to produce neutrophil-recruiting chemokines and antimicrobial factors such as ß-defensins, which may be relevant for staphylococcal defense (51). STAT3-deficient neutrophils display normal functions (52), but are prone to undergo S. aureus-induced cell death (53). Furthermore, STAT3-HIES patients display variable antibody responses and low numbers of memory B cells, which likely contributes to enhanced incidence of respiratory infections with H. influenzae and S. pneumoniae (52). STAT3 is ubiquitously expressed and multisystemic features are present. Thus, deficient epithelial STAT3 signaling may contribute to aberrant staphylococcal control by cytokine dysregulation and aberrant tissue remodeling (54, 55). STAT3 is involved in both pro- and anti-inflammatory signaling which complicates our understanding of single factors for the overall phenotype.
Autosomal-recessive ZNF341 deficiency leads to reduced cytokine signaling via STAT3 and resembles STAT3-HIES by displaying similar multisystemic features (e.g., bone fractures, retention of primary teeth, facial dysmorphism) but also staphylococcal infections (56).
IEI affecting single cytokines may teach us about their individual contribution. Lack of functional IL-6 cytokine family signaling reduces typical local inflammatory reaction, leads to low CRP and reduced systemic symptoms although tissue damage may be considerable. Defective IL-6 signaling either by IL-6 receptor deficiency (57) or by partial IL-6 signal transducer deficiency (IL6ST) (58) also leads to pyogenic infections, cold abscesses and pulmonary S. aureus infections. Additionally, phenocopies of IEI such as autoantibodies against IL-6 show increased susceptibility to S. aureus infection lacking CRP response (59). Staphylococcus aureus infections are also described in ERBIN deficiency which recapitulates some features of STAT3 deficiency (60).
Frequent S. aureus skin infections have also been reported in patients with STAT1GOF who are very susceptible to fungal infections, have low Th17 cells, and display a high rate of autoimmune features (61, 62).
IEI with defects in toll-like receptor (TLR)-signaling and susceptibility to S. aureusAutosomal-recessive IRAK-4 and MyD88 deficiencies affect TLR and IL-1R induced activation of NF-κB and MAPKs through the classical pathway (63). They disrupt key pathways in the innate immune response and usually present with bacterial pyogenic infections early in life (<2years of age). Most common pathogens are S. pneumoniae, S. aureus and Pseudomonas aeruginosa (64). Lack of TLR-induced signaling affects particularly the production of IL-6 and IL-8, and may lead to severe invasive infections (e.g., meningitis, sepsis, osteomyelitis, arthritis and abscesses), but also localized skin infections, lymphadenitis and ENT infections, usually without marked fever or increase of CRP (64). Still, pus is seen at the site of infection, which underlines that pus formation is not dependent on TLR-related cytokine signaling. As signs of infections may be absent but invasive infection may be rapidly progressing, it is vital to initiate empirical antibiotic treatment as soon as infection is suspected (64).
NEMO deficiency and IκBα GOF, which affect both NF-κB and TRIF-dependent signaling, result in a broad spectrum of immune dysfunctions and present also typically with colitis and ectodermal dysplasia. Apart from pyogenic bacterial infections, patients may also display mycobacterial infections, severe viral infections and opportunistic infections (64). Recently, more rare genetic defects associated to TLR-signaling have been reported, with variable phenotype depending on the protein involved.
Other diseases with susceptibility to S. aureusApart from classical IEI, increased susceptibility to S. aureus infections has also been reported in diseases such as cystic fibrosis, HIV infection and/or diabetes mellitus (65–68). In addition to aberrant host immune response, susceptibility to S. aureus may also be enhanced by colonization of multi-resistant strains (MRSA) carrying specific virulence factors.
Discussion: controversies, current knowledge gaps and future perspectivesWhile the key role of innate immunity for staphylococcal defense is well-established, the contribution of adaptive immunity is less clear.
In regards to B-cell immunity, evidence for a protective role of S. aureus antibodies is scarce. In fact, it has lately been suggested that S. aureus may induce non-protective antibodies, which then interfere with protective immune responses (69) facilitating commensalism and recurrent infections. Furthermore, patients with antibody deficiency do not display a specifically enhanced susceptibility to S. aureus, while they are clearly susceptible to other bacteria with a polysaccharide capsule (e.g., S. pneumoniae, H. influenzae). In contrast to the successful vaccine development for other encapsulated bacteria, there is still no available vaccine against S. aureus, and even adequate antibody induction to relevant S. aureus virulence factors did not lead to protection (70). The ability of anti-TSST-1 antibodies to provide protective immunity against superantigen-driven toxic shock syndrome appears to be an exception to the above, with IVIG being used as potential adjunctive therapy to ameliorate the symptoms (71).
Regarding the relevance of T cells, Th17 cells are often suggested to contribute to anti-staphylococcal-response, particularly at mucosa and skin sites (51). In mice, several studies document the importance of functional IL-17 signaling for the protection against mucocutaneous S. aureus infections (72, 73). Patients with IL-17RA deficiency are very prone to mucocutaneous candidiasis but do also display staphylococcal skin infections (74, 75). The initial hypothesis regarding the relevance of Th17 cells to prevent staphylococcal skin infection is closely related to the observed lack of Th17 in STAT3 deficiency (51). While the role of Th17 for candida defense is supported by other IEI with specifically deficient IL-17 signaling such as IL-17 autoantibodies (75), their relevance for S. aureus infections appears less significant. In the context of STAT3-HIES, the abundant changes in different cytokine signaling pathways and the contribution of ubiquitously deficient STAT3 needs to be considered. Of note, deficient IL-6 cytokine signaling is sufficient to predispose to staphylococcal infection even in the setting of normal Th17 cells (58, 76), and mere lack of Th17 cells does not induce susceptibility to S. aureus infection as evidenced in patients with IL12B/IL12RB1 deficiency (77) or CARD9 deficiency (78). Notably, STAT3-deficient patients with somatic mosaicism and normal Th17 compartment may still present with boils and pneumonia (79). Thus, lack of IL-17 signaling alone is likely insufficient in explaining enhanced susceptibility to S. aureus, even though patients may be more prone to folliculitis (74).
IEI with impairments in TLR and NF-κB signaling pathways such as in IRAK-4 or MyD88 deficiency, underline the significance of these pathways in recognizing and responding to S. aureus (80). Patients with STAT3-HIES, ZNF341 deficiency, partial IL6ST deficiency and IL-6 receptor deficiency all share deficient IL-6 signaling and enhanced frequency of “cold” staphylococcal abscesses and lung infections (1). IL-6 is a pleiotropic cytokine that is vital for acute-phase responses, defense against bacterial infections and tissue regeneration (81). The shared phenotype argues for an essential role of IL-6 in staphylococcal defense (82). Still, the precise molecular mechanism behind this particular predisposition and the contribution of other pathways is unknown.
Complement deficiencies might serve as additional risk factors in the context of S. aureus infections due to the crucial role of the complement system in opsonizing pathogens and facilitating their clearance by phagocytes. Susceptibility to S. aureus infections has been described in patients with C2 and C3 deficiencies (83) and complement activation was found to reduce persistent intracellular S. aureus burden in keratinocytes (84). Still, the role of complement in the defense against this pathogen appears less pronounced compared to its critical function in combating other encapsulated bacteria.
More recently, it has been proposed that specific genes may predispose to more severe infections via impairment of selective immune defense mechanism such as the altered response of non-leukocytic cells to staphylococcal alpha-toxin in OTULIN haploinsufficiency (85). With the growing use of NGS our understanding of specific factors in staphylococcal immunity will likely expand further. Still, the rareness of single IEI may hamper reliability of certain genotype-phenotype associations. An example is TYK2 deficiency, where the originally identified patient with susceptibility to S. aureus and hyper-IgE phenotype (86) was later judged to display deficient IL-6 signaling unrelated to TYK2 deficiency (87).
Last, the ability of S. aureus to survive intracellularly, notably within neutrophils, macrophages and as small colony variants in epithelial cells, complicates the immune response and treatment strategies and might facilitate recurrent infections (88). Together with the multiple other evasion strategies this poses significant challenges in vaccine development against S. aureus. In the light of growing rates of MRSA, it therefore remains essential to continue to assess host-pathogen interactions on a functional level and further enhance our understanding about crucial immune defense mechanisms.
Conclusion and diagnostic suggestions➢ Basic immunological workup in patients with recurrent or severe S. aureus infections should include a differential blood count and IgG, IgA, IgM, IgE
➢ Specific testing for CGD, HIES, complement deficiency, LAD, TLR deficiency, exclusion of secondary immunodeficiencies and assessment for phenocopies of IEI as well as genetic analysis may be warranted
➢ Inconclusive immunological investigation should be complemented by assessment of staphylococcal colonization
Author contributionsHK: Visualization, Writing – original draft, Writing – review & editing. KL: Writing – review & editing, Funding acquisition, Resources. SF: Writing – review & editing, Conceptualization, Supervision, Visualization, Writing – original draft, Methodology.
FundingThe author(s) declare financial support was received for the research and/or publication of this article.
This study was supported by the University Medical Center Hamburg-Eppendorf.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1389650/full#supplementary-material
AbbreviationsAD, autosomal-dominant; AIN, autoimmune neutropenia; AR, autosomal-recessive; C2, C3, complement component 2, complement component 3; CARD9, caspase recruitment domain family member 9; CD40, cluster of differentiation 40; CD40l, cluster of differentiation 40 ligand; CGD, chronic granulomatous disease; CID, combined immunodeficiency; CRP, C-reactive protein; DOCK, dedicator of cytokinesis; G-CSF, granulocyte colony-stimulating factor; GOF, gain of function; HAX1, HCLS1 associated protein X-1; HIES, hyper IgE syndrome; HIV, human immunodeficiency virus; HSCT, hematopoietic stem cell transplantation; IEI, inborn errors of immunity; Ig, immunoglobulin; IL, interleukin; IL-17RA, IL-17 receptor A; IL12B, IL12RB1, IL-12 subunit beta, IL-12 receptor beta 1; IL6ST, IL-6 signal transducer; IRAK-4, IL-1 receptor-associated kinase 4; IVIG, intravenous immunoglobulin; JAK, Janus kinase; LAD, leukocyte adhesion deficiency; LOF, loss of function; MAPK, mitogen-activated protein kinase; MRSA, methicillin-resistant Staphylococcus aureus; MyD88, myeloid differentiation primary response 88; NADPH, nicotinamide adenine dinucleotide phosphate; NEMO, NF-κB essential modulator; NF-kB, nuclear factor kappa B; NGS, next generation sequencing; Nox2, NADPH oxidase 2; OTULIN, OTU deubiquitinase with linear linkage specificity; PGM, phosphoglucomutase; PVL, panton-valentine leucocidin; RAC2, ras-related C3 botulinum toxin substrate 2; ROS, reactive oxygen species; S. aureus, Staphylococcus aureus; SCID, severe combined immunodeficiency; SCN, severe congenital neutropenia; SpA, staphylococcal protein A; spp, species pluralis; STAT3, signal transducer and activator of transcription 3; STAT1, signal transducer and activator of transcription 1; Th17, T helper 17; TLR, toll-like receptor; TMP/SMX, trimethoprim/sulfamethoxazole; TRIF, TIR-domain-containing adapter-inducing interferon-β; TSST-1, toxic shock syndrome toxin-1; TYK2, tyrosine kinase 2; ZNF341, zinc finger protein 341.
References1. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42(7):1473–507. doi: 10.1007/s10875-022-01289-3
PubMed Abstract | Crossref Full Text | Google Scholar
2. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. (2015) 28(3):603–61. doi: 10.1128/CMR.00134-14
PubMed Abstract | Crossref Full Text | Google Scholar
3. Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. (2005) 5(12):751–62. doi: 10.1016/S1473-3099(05)70295-4
PubMed Abstract | Crossref Full Text | Google Scholar
4. Kwiecinski JM, Horswill AR. Staphylococcus aureus bloodstream infections: pathogenesis and regulatory mechanisms. Curr Opin Microbiol. (2020) 53:51–60. doi: 10.1016/j.mib.2020.02.005
PubMed Abstract | Crossref Full Text | Google Scholar
6. David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev. (2010) 23(3):616–87. doi: 10.1128/CMR.00081-09
PubMed Abstract | Crossref Full Text | Google Scholar
8. Wójcik-Bojek U, Różalska B, Sadowska B. Staphylococcus aureus-a known opponent against host defense mechanisms and vaccine development-do we still have a chance to win? Int J Mol Sci. (2022) 23(2). doi: 10.3390/ijms23020948
Crossref Full Text | Google Scholar
9. de Jong NWM, van Kessel KPM, van Strijp JAG. Immune evasion by Staphylococcus aureus. Microbiol Spectr. (2019) 7(2).30927347
PubMed Abstract | Google Scholar
10. Loh JM, Aghababa H, Proft T. Eluding the immune system’s frontline defense: secreted complement evasion factors of pathogenic gram-positive cocci. Microbiol Res. (2023) 277:127512. doi: 10.1016/j.micres.2023.127512
PubMed Abstract | Crossref Full Text | Google Scholar
12. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. (2014) 60(8):1176–83. doi: 10.1093/cid/ciu1154
PubMed Abstract | Crossref Full Text | Google Scholar
13. van den Berg JM, Kuijpers TW. Educational paper: defects in number and function of neutrophilic granulocytes causing primary immunodeficiency. Eur J Pediatr. (2011) 170(11):1369–76. doi: 10.1007/s00431-011-1584-5
PubMed Abstract | Crossref Full Text | Google Scholar
14. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. (2010) 2(6):576–86. doi: 10.1159/000319909
PubMed Abstract | Crossref Full Text | Google Scholar
15. Löffler B, Tuchscherr L, Niemann S, Peters G. Staphylococcus aureus persistence in non-professional phagocytes. Int J Med Microbiol. (2014) 304(2):170–6. doi: 10.1016/j.ijmm.2013.11.011
Crossref Full Text | Google Scholar
17. Missiakas D, Winstel V. Selective host cell death by Staphylococcus aureus: a strategy for bacterial persistence. Front Immunol. (2020) 11:621733. doi: 10.3389/fimmu.2020.621733
PubMed Abstract | Crossref Full Text | Google Scholar
19. Kobayashi SD, DeLeo FR. Staphylococcus aureus protein A promotes immune suppression. mBio. (2013) 4(5). doi: 10.1128/mbio.00764-13
Crossref Full Text | Google Scholar
20. Miethke T, Duschek K, Wahl C, Heeg K, Wagner H. Pathogenesis of the toxic shock syndrome: t cell mediated lethal shock caused by the superantigen TSST-1. Eur J Immunol. (1993) 23(7):1494–500. doi: 10.1002/eji.1830230715
PubMed Abstract | Crossref Full Text | Google Scholar
22. Han X, Lu S, Gu C, Bian Z, Xie X, Qiao X. Clinical features, epidemiology, and treatment of Shwachman-Diamond syndrome: a systematic review. BMC Pediatr. (2023) 23(1):503. doi: 10.1186/s12887-023-04324-3
PubMed Abstract | Crossref Full Text | Google Scholar
23. Bux J, Behrens G, Jaeger G, Welte K. Diagnosis and clinical course of autoimmune neutropenia in infancy: analysis of 240 cases. Blood. (1998) 91(1):181–6. doi: 10.1182/blood.V91.1.181
PubMed Abstract | Crossref Full Text | Google Scholar
24. Farruggia P, Dufour C. Diagnosis and management of primary autoimmune neutropenia in children: insights for clinicians. Ther Adv Hematol. (2015) 6(1):15–24. doi: 10.1177/2040620714556642
PubMed Abstract | Crossref Full Text | Google Scholar
25. Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause KH. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol Rev. (2017) 41(2):139–57.27965320
PubMed Abstract | Google Scholar
26. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. (2009) 4(4):e5234. doi: 10.1371/journal.pone.0005234
PubMed Abstract | Crossref Full Text | Google Scholar
28. Slack MA, Thomsen IP. Prevention of infectious complications in patients with chronic granulomatous disease. J Pediatric Infect Dis Soc. (2018) 7(suppl_1):S25–30. doi: 10.1093/jpids/piy016
PubMed Abstract | Crossref Full Text | Google Scholar
29. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. (2010) 116(9):1570–3. doi: 10.1182/blood-2010-01-264218
PubMed Abstract | Crossref Full Text | Google Scholar
31. Roos D, van Leeuwen K, Madkaikar M, Kambli PM, Gupta M, Mathews V, et al. Hematologically important mutations: leukocyte adhesion deficiency (second update). Blood Cells Mol Dis. (2023) 99:102726. doi: 10.1016/j.bcmd.2023.102726
PubMed Abstract | Crossref Full Text | Google Scholar
32. Gazit Y, Mory A, Etzioni A, Frydman M, Scheuerman O, Gershoni-Baruch R, et al. Leukocyte adhesion deficiency type II: long-term follow-up and review of the literature. J Clin Immunol. (2010) 30(2):308–13. doi: 10.1007/s10875-009-9354-0
PubMed Abstract | Crossref Full Text | Google Scholar
33. Banday AZ, Nisar R, Patra PK, Kaur A, Sadanand R, Chaudhry C, et al. Clinical and immunological features, genetic variants, and outcomes of patients with CD40 deficiency. J Clin Immunol. (2023) 44(1):17. doi: 10.1007/s10875-023-01633-1
PubMed Abstract | Crossref Full Text | Google Scholar
34. Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A. (2001) 98(22):12614–9. doi: 10.1073/pnas.221456898
PubMed Abstract | Crossref Full Text | Google Scholar
35. Ferrua F, Galimberti S, Courteille V, Slatter MA, Booth C, Moshous D, et al. Hematopoietic stem cell transplantation for CD40 ligand deficiency: results from an EBMT/ESID-IEWP-SCETIDE-PIDTC study. J Allergy Clin Immunol. (2019) 143(6):2238–53. doi: 10.1016/j.jaci.2018.12.1010
PubMed Abstract | Crossref Full Text | Google Scholar
37. Cabral-Marques O, França TT, Al-Sbiei A, Schimke LF, Khan TA, Feriotti C, et al. CD40 ligand deficiency causes functional defects of peripheral neutrophils that are improved by exogenous IFN-γ. J Allergy Clin Immunol. (2018) 142(5):1571–88.9. doi: 10.1016/j.jaci.2018.02.026
PubMed Abstract | Crossref Full Text | Google Scholar
38. Hsu AP, Donkó A, Arrington ME, Swamydas M, Fink D, Das A, et al. Dominant activating RAC2 mutation with lymphopenia, immunodeficiency, and cytoskeletal defects. Blood. (2019) 133(18):1977–88. doi: 10.1182/blood-2018-11-886028
PubMed Abstract | Crossref Full Text | Google Scholar
39. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. (2000) 97(9):4654–9. doi: 10.1073/pnas.080074897
PubMed Abstract | Crossref Full Text | Google Scholar
40. Donkó A, Sharapova SO, Kabat J, Ganesan S, Hauck F, Marois L, et al. Clinical and functional spectrum of RAC2-related immunodeficiency. Blood. (2024).
41. Fallahi M, Jamee M, Enayat J, Abdollahimajd F, Mesdaghi M, Khoddami M, et al. Novel PGM3 mutation in two siblings with combined immunodeficiency and childhood bullous pemphigoid: a case report and review of the literature. Allergy Asthma Clin Immunol. (2022) 18(1):111. doi: 10.1186/s13223-022-00749-0
PubMed Abstract | Crossref Full Text | Google Scholar
42. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. (2009) 361(21):2046–55. doi: 10.1056/NEJMoa0905506
PubMed Abstract | Crossref Full Text | Google Scholar
43. Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. (2009) 124(6):1289–302.4. doi: 10.1016/j.jaci.2009.10.038
PubMed Abstract | Crossref Full Text | Google Scholar
44. Wilkie H, Das M, Pelovitz T, Bainter W, Woods B, Alasharee M, et al. Regulatory T-cell dysfunction and cutaneous exposure to Staphylococcus aureus underlie eczema in DOCK8 deficiency. J Allergy Clin Immunol. (2024).38185418
PubMed Abstract | Google Scholar
45. Wilkie H, Timilshina M, Rahmayanti S, Das M, Pelovitz T, Geha RS. DOCK8 is essential for neutrophil mediated clearance of cutaneous S. aureus infection. Clin Immunol. (2023) 254:109681. doi: 10.1016/j.clim.2023.109681
PubMed Abstract | Crossref Full Text | Google Scholar
46. Keles S, Charbonnier LM, Kabaleeswaran V, Reisli I, Genel F, Gulez N, et al. Dedicator of cytokinesis 8 regulates signal transducer and activator of transcription 3 activation and promotes T(H)17 cell differentiation. J Allergy Clin Immunol. (2016) 138(5):1384–94.2. doi: 10.1016/j.jaci.2016.04.023
PubMed Abstract | Crossref Full Text | Google Scholar
48. Farmand S, Sundin M. Hyper-IgE syndromes: recent advances in pathogenesis, diagnostics and clinical care. Curr Opin Hematol. (2015) 22(1):12–22. doi: 10.1097/MOH.0000000000000104
PubMed Abstract | Crossref Full Text | Google Scholar
49. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. (2008) 452(7188):773–6. doi
留言 (0)