The incidence of AD, a leading cause of disability and death, is expected to grow rapidly over the next decade [1]. The financial, physical, and emotional burden placed on patients and caregivers can be overwhelming [2]. Patient's experiences irreversible dementia and increasing loss of cognition, eventually becoming entirely dependent on caretakers in everyday life. Most available treatments are limited to symptomatic benefits: cholinesterase inhibitors are used to improve cognition, anti-depressants treat dementia-related depression, and various pain medications may be used to increase patient comfort [[3], [4], [5]]. Decades of research have shown that AD's progression involves the build-up of amyloid-beta (Aβ)-deposits (plaques), consisting of insoluble peptide fragments (Aβ1-42) and tau protein aggregation (Neurofibrillary tangles (NFTs) in the brain [6]. The vast majority of disease-modifying therapies are developed to reduce these plaques and NFTs [7]. This approach has yielded two approved disease-modifying drug Aducanumab and Lecanemab, whose efficacy is currently still debated [8]. Both these drugs are monoclonal antibodies (MAb) in nature and have shown to reduce the build-up of plaques containing the Aβ peptide, but not associated with cognitive impairment or other important outcomes [9]. As such, new drug targets are needed to pave the way forward for more efficacious therapeutics.
The exact cause of AD is still unknown owing to the multifaceted pathophysiology involved in the development and progression of the disease. Though several hypothesis have been proposed that includes, the cholinergic hypothesis that governs the lower acetylcholine (ACh) levels in the synaptic cleft, Central nervous system (CNS) inflammation in response to activated microglial cells and astrocytes, activation of N-methyl-d-aspartate receptor (NMDAR), Oxidative stress, cyclic-AMP-response element-binding protein (CREB) signaling pathways, apolipoprotein E4 (APOε4) gene transcription, accumulation of amyloid beta (Aβ) and its aggregates and hyperphosphorylated tau and formation of neurofibrillary tangles (NFTs) etc [[10], [11], [12], [13], [14], [15]]. However, last two hypotheses are the most acceptable amongst several proposed hypotheses for AD development and progression.
Dyrk1A, a protein kinase and is reported to be overexpressed in AD brain, in reactive astrocytes and microglia, cell types contributing to the pathology and progression of AD [16,17]. Genetic studies suggest an association of Dyrk1A with neurodegeneration, Aβ-deposition and dementia, especially in Down's syndrome and Parkinson's disease [[18], [19], [20]]. Dyrk1A was found to be abnormally expressed in post-mortem AD brain slices, notably increased in reactive astrocytes lining and Aβ plaques [21]. Dyrk1A has been shown to phosphorylate known AD proteins: amyloid precursor protein (APP), tau protein, and presenilin 1 [[22], [23], [24]]. In animal models of AD, Dyrk1A inhibitors improve amyloid pathology, motor function, memory, and most notably, neuroinflammation, a process associated closely with the activation of astrocytes and microglia [[25], [26], [27], [28]]. An understanding of how Dyrk1A is related to these pathologies will provide further validation of this protein as a drug target in AD.
BACE-1 also promotes Aβ aggregation by catalytic proteolysis of APP, which results in the formation of monomers, protofibrils, annular oligomers, fibrils, and plaques. Aβ aggregates may form free radicals in the mitochondria and lead to oxidative stress and neurodegeneration [29]. Therefore, the multitargeting single ligand approach may be a promising strategy than selectively targeting enzyme to overcome the disease rather than providing symptomatic relief in AD.
The idea behind the present work was to design multitargeted directed ligands (MTDLs) for the treatment of AD utilizing a molecular hybridization and in silico design approaches. Three targets, namely Cholinesterases (ChE), Dyrk1A and BACE-1 are considered in this work to design a MTDL inhibitor molecule. These targets are mainly responsible for the development of Aβ aggregation and NFTs leading to neurodegeneration [[30], [31], [32]]. AChE-PAS activates APP through the amyloidogenic pathway and triggers Aβ aggregation [33]. While, BACE-1 has direct involvement through the amyloidogenic pathway by activating APP in developing Aβ aggregation [34]. Tau and APP hyperphosphorylation through Dyrk1A is believed to trigger the formation of NFTs and Aβ-aggregation. This hyperphosphorylation also induces the release of some inflammatory cytokines and activates astrocytes producing neurotoxicity [35]. The Aβ aggregation and formation of NFTs disrupt cellular and molecular communication characterized by memory and behavioral deficits in the AD brain [36]. Thus by inhibiting all these targets togather will enhance symtomatic functions and retard AD progression.


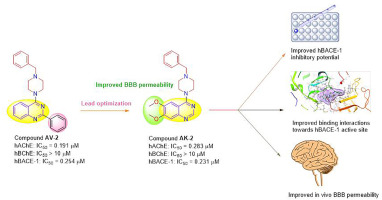




留言 (0)