Inflammatory bowel disease (IBD), primarily Crohn's disease and ulcerative colitis (UC), is a chronic, intermittent condition that affects the gastrointestinal system and can be life-threatening. It is characterized by extensive enteric inflammation [1]. The development of UC is believed to involve a complex interplay of factors such as immune dysregulation, genetic susceptibility, excessive inflammatory response, endoplasmic reticulum (ER) stress, impaired epithelial barrier, and alterations in the intestinal flora microbiota. Despite ongoing research, the exact mechanisms underlying UC remain poorly understood [2].
Oxazolone (OXZ)-induced colitis is now the most extensively studied animal model of T helper-2 (Th-2)-mediated immune activation [3]. OXZ can activate Th-2 and invariant natural killer T cells, resulting in a significant increase in interleukin 13 (IL-13) and other cytokines like IL-4 and IL-5. Elevated IL-4 levels stimulate Th-2 cells to produce more IL-4, intensifying the inflammatory response via positive feedback. Notably, evidence suggests that OXZ induces a typical presentation of UC, primarily affecting the distal colon, specifically the mucosal and submucosal layers [4]. The histopathological findings and Th-2 cytokine production in OXZ-induced colitis are consistent with those reported during the active phase of human UC [5].
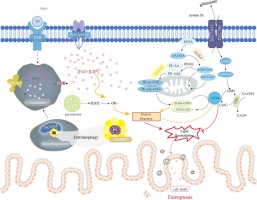
Numerous lines of research support the hypothesis that mitochondria play an important role in cellular physiology during UC development [6]. Interestingly, mitochondrial dysfunction, which is primarily caused by mtDNA mutations, increased reactive oxygen species (ROS) generation, and decreased adenosine triphosphate (ATP) generation, is linked to gastrointestinal tract dysfunction and even enteric inflammation [7]. Additionally, growing evidence suggests that mitochondrial abnormalities in colitis may be responsible for epithelial barrier breakdown. According to Ho et al.'s [8] findings, loss of multidrug resistance-1 causes mitochondrial dysfunction and increased mtROS generation, resulting in epithelial barrier defects in colitis.
Autophagy is a crucial biological process that maintains cellular homeostasis by sequestering senescent or damaged organelles or proteins in autophagosomes, which are then lysed in lysosomes to recycle their components [9]. Several chronic inflammatory diseases, including IBD, are associated with impaired autophagy. Autophagy plays a critical role in IBD etiopathogenesis by altering processes such as intracellular bacterial killing, Paneth cell antimicrobial peptide release, goblet cell function, macrophage cytokine release, dendritic cell antigen presentation, and enterocyte ER stress response [10]. Therefore, a better understanding of autophagy's role in UC prevention and treatment can help to optimize existing UC treatment strategies.
Intriguingly, there is strong evidence that adenosine monophosphate-activated protein kinase (AMPK) modulates inflammatory responses in several animal models of inflammation, and AMPK agonists have previously been shown to control inflammatory responses in IBD [11]. Recent studies suggest that activating AMPK may facilitate the nuclear translocation of nuclear factor erythroid 2 p45-related factor 2 (Nrf2), a crucial regulator of redox homeostasis [12]. Upon nuclear translocation, it binds to antioxidant response elements in genes that encode detoxification and antioxidant enzymes, enhancing transcription and exerting antioxidant activity [13]. Nrf2 also has a substantial influence on mitochondrial function [14].
Notably, the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and mammalian target of rapamycin (mTOR) signaling pathways play a crucial role in regulating and releasing inflammatory cytokines that contribute to UC development [15], [16]. These pathways also affect oxidative stress (OS), autophagy, apoptosis, and cell proliferation [17]. Additionally, mTOR regulates ER stress [18]. Thus, targeting this pathway could lead to innovative therapeutics for managing UC or halting its progression.
A significant class of oral glucose-lowering agents are dipeptidyl peptidase IV (DPP-IV) inhibitors, which inhibit the breakdown of glucagon-like peptides [19]. Previous studies have shown the efficacy of DPP-IV inhibitors in promoting intestinal healing by resolving mucosal damage and inflammation [20]. Vildagliptin (Vilda), a potent and selective DPP-IV inhibitor, offers numerous beneficial effects, including anti-inflammatory, anti-apoptotic, and antioxidative properties [21]. Additionally, Vilda has been reported to modulate autophagy [22]. Although Vilda has recently been reported to alleviate acetic acid-induced UC [21], more research is needed to elucidate the mechanisms underlying Vilda’s protective effects on the colon.
A definitive cure for UC remains elusive, and current treatment options are limited to anti-inflammatory and immunomodulatory medications. Therefore, there is an urgent need for innovative, safe, and effective therapies for UC. Hence, this study aimed to explore the potential coloprotective efficacy of Vilda in an OXZ-induced UC model and to identify the molecular pathways involved. To the best of our knowledge, this is the first study investigating Vilda's therapeutic potential against OXZ-induced UC in a rat model. This study emphasizes Vilda's potential impact on ameliorating colonic inflammation, oxidative/ER stress, mitochondrial dysfunction, and promoting autophagy via the PI3K/AKT/mTOR and AMPK/Nrf2 signaling pathways.








留言 (0)