
記住我

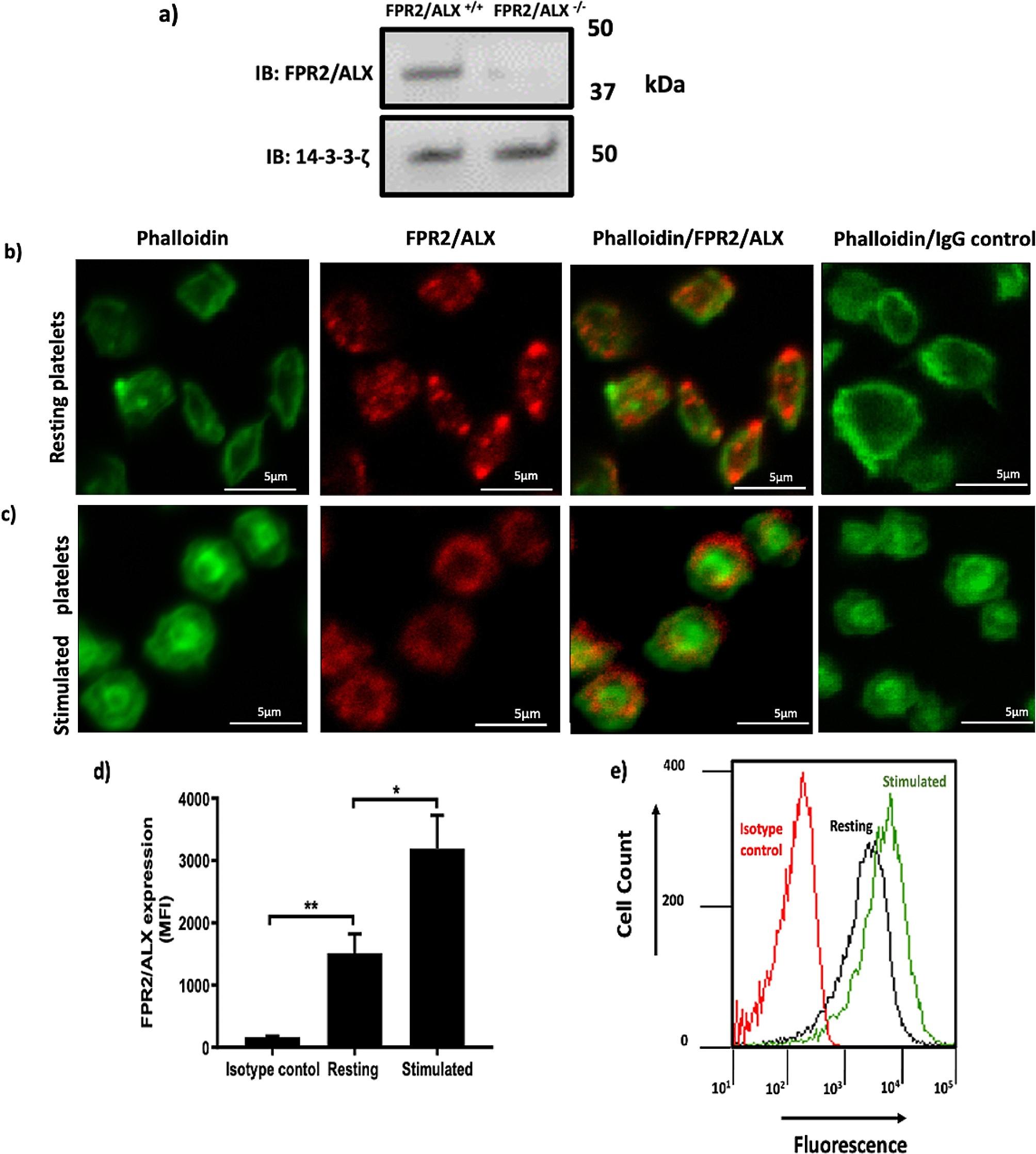

The presence of FPR2/ALX mRNA in megakaryocytes highlighted the expression of this receptor in human and mouse platelets [8, 12]. In addition, the expression of FPR2/ALX in human and mouse platelets has been previously confirmed at protein levels through immunoblot analysis [8, 13], however, the distribution of the receptor in human platelets has not been addressed yet. Therefore, we investigated the subcellular localisation of the receptor in permeabilised-resting platelets with the aid of anti-FPR2/ALX antibody. The specificity of the antibody was confirmed on FPR2/ALX-deficient platelet as shown in Fig. 1a. Using immunofluorescence microscopy, FPR2/ALX was observed around the periphery of platelets in resting condition. Interestingly, FPR2/ALX was also seen inside the cytosol of platelets (Fig. 1b). However, upon activation, FPR2/ALX seems to get mobilised towards the platelet periphery (Fig. 1c ). This finding was further confirmed using flow cytometry. Human PRP was stimulated with vehicle control or 0.5 µg/ml CRP-XL prior to the addition of anti-FPR2/ALX antibody and incubation for another 10 min. The secondary antibody (Cy5™ conjugated anti-rabbit IgG) was added and incubated for a further 10 min. Samples were then fixed with 0.2% (v/v) formyl saline, and the level of fluorescence was measured using a flow cytometer. In agreement with the imaging findings, variation in the distribution of FPR2/ALX receptors in resting and stimulated platelets was also noted (Fig. 1d, e).
Fig. 1
Immunofluorescence and flow cytometry analyses of resting and stimulated platelets confirm the presence of FPR2/ALX. (a) FPR2/ALX+/+ and FPR2/ALX−/− mouse platelet were lysed and analysed by SDS-PAGE. Following blotting of the separated proteins on a PVDF membrane, the primary (rabbit polyclonal anti-FPR2/ALX antibody) and secondary (Cy5™ anti-rabbit IgG antibody) antibodies were used to detect FPR2/ALX. 14-3-3-ζ was included as a loading control. The membrane was scanned using a Typhoon 9400 Variable Mode Imager (GE Healthcare, UK) to visualise the bands. (b) Resting and (c) 5 µM U46619-stimulated platelets were fixed and permeabilised by using 0.2% (V/V) TritonTM x-100 before the addition of anti-FPR2/ALX antibody and Alexa Fluor 488-conjugated Phalloidin. Alexa Fluor 647-labelled anti-rabbit IgG antibodies were added to detect FPR2/ALX. As controls, Alexa Fluor 647-anti-rabbit IgG and Phalloidin were added. Negative controls excluded the non-specific binding. Platelets were then visualised by confocal microscopy, objective (1000x). The images shown are representative of several images taken for three separate donors. Human PRP [resting or CRP-XL (0.5 µg/ml) stimulated] was incubated with anti-FPR2/ALX antibodies and detected using Cy5TM anti-rabbit IgG secondary antibodies and analysed by flow cytometry. (d) The bar chart represents the surface expression of FPR2/ALX in platelets, and its expression increases compared to isotype control. (e) A representative histogram showing the elevation of FPR2/ALX levels in activated platelets (green) compared to resting platelets (black). The red line represents the isotype control. Data represent the mean ± SEM (n = 3) of the median fluorescence intensity values. *P ≤ 0.05 and **P ≤ 0.01 values were as calculated by One-way ANOVA
BML-111 inhibits platelet aggregationBML-111 is a LXA4 analogue that has been proven to selectively bind FPR2/ALX receptors in platelet with the aid of FPR2/ALX-deficient platelets (supplementary Fig. 1a &b). We studied the impact of BML-111 on platelet aggregation following stimulation with crosslinked collagen-related peptide (CRP-XL), a selective GPVI agonist, and thrombin, an agonist for protease-activated receptors (PARs). Human isolated platelets were incubated with either vehicle control (modified-Tyrode’s HEPES buffer) or a range of different concentrations of BML-111 for 5 min prior to the addition of CRP-XL or thrombin, the tested BML concentrations were chosen based on a toxicity assay (supplementary Fig. 1c). The aggregation was monitored under stirring conditions at 37 °C for 300 s using light transmission aggregometry (Fig. 2). As shown in Fig. 2a and b, pre-treatment with BML-111 inhibited platelet aggregation in a dose-dependent manner when compared to the vehicle-treated sample in response to CRP-XL stimulation. Similarly, thrombin-induced platelet aggregation was also inhibited in the presence of increasing concentrations of BML-111 (Fig. 2c and d).
BML-111 reduces inside-out signaling to integrin αIIbβ3 and platelet degranulationCrosslinking of platelets occurs through the binding of fibrinogen to activated integrin αIIbβ3 that leads to the formation of platelet aggregates. Given the inhibitory effects of BML-111 on platelet aggregation, we sought to investigate the influence of this ligand on integrin αIIbβ3 activation using FITC-conjugated rabbit anti-human fibrinogen antibody via flow cytometry. Fibrinogen binding to CRP-XL stimulated platelets was reduced by around 35%, 49%, 57% and 64% at 6.25, 12.5, 25 and 50 µM of BML-111, respectively (Fig. 2e). The inhibitory effect of BML-111 was also observed in thrombin stimulated platelets at higher concentrations of BML-111 (Fig. 2f). These findings are in alignment with inhibited platelet aggregation upon stimulation with CRP-XL or thrombin following ligation of FPR2/ALX with BML-111. In addition, the impact of BML-111 on the α-granule secretion of platelets was analysed using PE-Cy5 conjugated mouse anti-human CD62P antibody by flow cytometry. As shown in Fig. 2, significant and dose-dependent inhibition of P-selectin exposure was observed when different concentrations of BML-111 were added upon stimulation with CRP- XL (Fig. 2g). Moreover, thrombin mediated exposure of P-selectin was significantly reduced when platelets were treated with BML-111 (Fig. 2h). Also, we measured the effect of BML-111 on ATP release from activated platelets using a luciferin-luciferase reagent. BML-111 treatment negatively impacted ATP release at various concentrations when stimulated with CRP-XL as demonstrated in Fig. 2i &j. Likewise, inhibition was also observed in thrombin stimulated platelets when compared to vehicle-treated samples as shown in Fig. 2k &l.
Fig. 2
BML-111 inhibits platelet aggregation, integrin αIIbβ3 activation and secretion. Human isolated platelets (4 × 108 cells/ml) were incubated with modified-Tyrode’s HEPES buffer or different concentrations of BML-111 (3.125, 6.25, 12.5, 25, and 50 µM) for 5 min and then stimulated with CRP-XL (0.25 µg/ml) and thrombin (0.05 U/ml). The change in light transmission was monitored and recorded for 300 s. (a, c) Representative aggregation traces from three separate donors. (b, d) Cumulative data from three different donors. The percentage of aggregation measured at 300 s with a vehicle control was taken as 100%. Human PRP was incubated with different concentrations of BML-111 (3.125, 6.25, 12.5 and 50 µM) or vehicle control (modified-Tyrode’s HEPES buffer) for 5 min prior to stimulating with (e) CRP-XL (0.25 µg/ml) or (f) thrombin (0.05U/ml) for 20 min followed by flow cytometry analysis. Integrin αIIbβ3 activation (inside-out signaling) was determined by measuring the level of fibrinogen binding to the platelets. The bar charts represent the percentage of fibrinogen binding compared with the positive control, which is defined as 100%. The level of P-selection exposure on the platelet surface was measured using anti-human CD62P antibodies by flow cytometry. The bar charts represent the percentage of P-selectin exposure compared with the positive control, which is defined as 100% (g, h). The level of ATP release was observed by lumi-aggregometery using a luciferin-luciferase detection system. (i, k) Representative traces for ATP release. (j, l) ATP secretion was calculated as a percentage of the area under the curve, where 100% was expressed as the level of ATP release achieved with a vehicle control at 300 s. Data represent mean ± SEM (n = 3). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001 values were as calculated by One-way ANOVA.
BML-111 negatively impacts integrin αIIbβ3 outside-in signalingPlatelet spreading and clot retraction occur due to outside-in signaling events mediated by integrin αIIbβ3 following fibrinogen binding to ensure the stability of the growing thrombus. To assess the impact of BML-111 on the outside-in signaling, platelet spreading on fibrinogen coated coverslips was evaluated in BML-111 treated platelets. 25 and 50 µM BML-111 significantly reduced platelet adhesion treatment by 62% and 73%, respectively. Also, approximately 80% of platelets in vehicle-treated samples showed a high degree of lamelliopodia. On the other hand, pre-treatment with 25 and 50 µM of BML-111 inhibited the levels of platelet spreading to fibrinogen coated coverslips (Fig. 3a, b &c). Similarly, clot retraction was impeded by BML-111 treatment as shown in Fig. 3d &e. These findings indicate a role of FPR2/ALX in the formation and stability of thrombi.
Fig. 3
BML-111 prevents adhesion and spreading of platelets on immobilised fibrinogen and clot retraction. Human isolated platelets (2 × 107 cells/ml) were incubated with modified-Tyrode’s HEPES buffer as vehicle control or different concentrations of BML-111 (12.5, 25, and 50 µM) for 5 min at room temperature and then dispensed onto glass coverslips coated with fibrinogen (100 µg/ml) and incubated at 37 °C for 1 h. Samples were fixed with 0.2% (w/v) formyl saline and then permeabilised with 0.2% (v/v) TritonTM x-100. Adhered platelets were stained with Alexa-Fluor 488 Phalloidin for 1 h in the dark and mounted onto glass slides. Platelets were then visualised using confocal microscopy, objective (100x). Multiple images were taken randomly for each slide. (a) Representative image of platelet adhesion and spreading on fibrinogen. (b) An average number of platelets adhered. (c) Spreading of platelets was divided into 3 types: adhered, filopodia and lamelliopodia. (d) PRP was incubated with modified-Tyrode’s HEPES buffer or different concentrations of BML-111 (3.125, 6.25, 12.5, 25 and 50 µM) along with 10 µl red blood cells in glass test tubes for 15 min at room temperature. Thrombin (1 U/ml) was added to initiate clot formation. The clot was retracted around a sealed glass capillary tube placed in the middle of the tube. Clots were observed over 90 min. (e) The clot retraction was calculated by measuring the clot weights. The cumulative data were used to show clot weight. Data represent mean ± SEM (n = 3). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001values were as calculated by One-way ANOVA.
BML-111 negatively impacts calcium mobilisationKnowing the important role of Ca2+ in several vital stages of platelet activation from inside-out signalling to degranulation, it is important to study the impact of BML-111 on intracellular Ca2+ mobilisation following CRP-XL or thrombin stimulation. A membrane permeable calcium dye, Fura-2AM was used to monitor the calcium levels through binding to free cytosolic Ca2+. As a result of Ca2+ binding, the dye gets excited at wavelength 340 and 380 nm and then emitted at 510 nm. Isolated platelets (4 × 108 cells/ml) loaded with Fura-2AM were incubated with BML-111 (25 and 50 µM) or a vehicle control (modified-Tyrode’s HEPES buffer) for 5 min prior to stimulation with CRP-XL (0.25 µg/mL) or thrombin (0.05 U/mL) for 5 min. Levels of Ca2+ mobilisation were assessed by measuring the fluorescence. BML-111 treatment with 25 and 50 µM in CRP-XL stimulated platelets displayed an inhibition in Ca2+ levels as reflected in cytosolic Ca2+mobilisation. BML-111 at 25 and 50 µM showed a significant reduction in cytosolic calcium levels by approximately 25% and 40%, respectively, in comparison to the vehicle control (Fig. 4b). Similarly, a concentration-dependent reduction in calcium mobilisation was observed in 25 and 50 µM BML-111 treated platelets of 37% and 57%, respectively, in comparison to vehicle control following thrombin-stimulation (Fig. 4d).
Fig. 4
BML-111 inhibits intracellular Ca2+ mobilisation. Fura-2 AM-loaded platelets (4 × 108 cells/ml) were treated with BML-111 (25 and 50 µM) or vehicle control (modified-Tyrode’s HEPES buffer) for 5 min, then stimulated with CRP-XL (0.25 µg/mL) or thrombin (0.05 U/mL) for 5 min. (a, c) Traces of cytosolic calcium mobilisation after stimulation with CRP-XL or thrombin. (b, d) Cumulative data of the peak intracellular Ca2+ levels were shown. Peak calcium levels achieved in the presence of vehicle control defines 100%. Fluorescent intensity was measured by a plate reader (with excitation at 340 and 380 nm and emission at 510 nm). Data represent the mean ± SEM (n = 4). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001 were as calculated by One-way ANOVA.
In vitro thrombus formation is reduced by BML-111Given the impact of BML-111 on several aspects of platelet activation, the work was extended to assess the effect of BML-111 on thrombus formation in vitro under arterial flow conditions. Human whole blood was incubated with a lipophilic dye, DiOC6 (5 µM) at 30 °C for 1 h. Then, dye labelled whole blood was incubated with BML-111 or a vehicle-control for 5 min prior to perfusion through the collagen-coated chip at an arteriolar shear rate of 20 dynes/cm2. Fluorescence was excited at 488 nm with an argon laser and emission was measured at 500–520 nm. Thrombus formation on the microfluidic chip was observed by using a Nikon A1R confocal microscope with a 20X objective. Representative images at the end of the assay show big, stable, and bright formed thrombi in the vehicle-control treated sample (Fig. 5a). On the other hand, treatment with BML-111 significantly reduced the size of the formed thrombi. Collectively, 25, and 50 µM BML-111 treatment caused a significant reduction in thrombus formation by approximately 43% and 50%, respectively (Fig. 5b).
Fig. 5
BML-111 attenuates thrombosis. A lipophilic dye, DiOC6 (5 µM) was added to citrated human blood and incubated for 1 h at 30 ℃, then treated with BML-111 (25 and 50 µM) or a vehicle-control (modified-Tyrode’s HEPES buffer) for 5 min. The labelled blood was then perfused across Vena8 Biochips, coated with collagen (100 µg/ml) under arterial flow conditions (shear stress: 20 dynes/cm2). (a) Representative images show thrombus formation over 540 s in 25 and 50 µM BMLL-111 or vehicle-control treated samples. Fluorescence was measured using an excitation wavelength of 488 nm and emission at 500–520 nm with an argon laser. A Nikon A1R confocal microscope (20x objective) was used to observe thrombus formation, and images were captured every second for over 600 s. (b) Quantified data represents mean thrombus fluorescence intensity for BML-111 and vehicle-control treated samples. The data were analysed using NIS-Elements software and normalised to the fluorescence level at the end of the assay in the vehicle-control treated sample. Data represent the mean ± SEM (n = 4), ****P ≤ 0.0001 was as calculated by Two-way ANOVA.
BML-111 upregulates inhibitory pathway-mediated signalingGi-coupled FPR2/ALX has been linked to the regulation of platelet inhibitory signaling pathways of cAMP and hence increasing platelet reactivity. Nevertheless, BML-111 demonstrated a potential down-regulation of platelet activity and thrombosis. To further explain its inhibitory effect, we looked into the impact of BML-111 treatment on the phosphorylation of VASP-S157 and VASP-S239, markers of cAMP/PKA and cGMP/PKG activity, respectively, in resting and stimulated platelets. To achieve maximum phosphorylation levels of VASP S-157 and S-239, isolated platelets were treated with positive controls, PGI2 (1 µg/ml) and PAPA-Nonoate (100 µM; NO donor), respectively. As shown in Fig. 6a, pre-treatment of platelets with 25 and 50 µM BML-111 caused a significant increase in VASP S-157 phosphorylation by 86% and 88%, respectively, when compared to vehicle-treated platelets in resting conditions. Similarly, stimulated platelets showed an increase of VASP S-157 phosphorylation when treated with 12.5, 25 and 50 µM BML-111 in a dose dependent manner (Fig. 6b). However, when compared to vehicle samples, no changes have been noted in the phosphorylation levels of VASP S-239 in BML-111 treated samples in resting and stimulated conditions (supplementary Fig. 2a &b).
Given the effects of BML-111 on VASP S-157 phosphorylation, we assessed the levels of cAMP in the presence of BML-111. Interestingly, cAMP levels were not increased upon BML-111 treatment (supplementary Fig. 3). However, the elevation of cAMP levels was indeed observed in PGI2 treated control sample suggesting that BML-111 modulates VASP S-157 phosphorylation independently of cAMP. Moreover, H-89 dihydrocholride hydrate (10 µM), a selective inhibitor of PKA [14], and SQ 22536 (100 µM), an AC blocker which inhibits its activity and prevents cAMP production [15], inhibited VASP S-157 phosphorylation in the presence of PGI2 confirming that PGI2-induced VASP S-157 phosphorylation is modulated via cAMP/PKA signaling pathway. On the contrary, BML-111- mediated increase in VASP S-157 phosphorylation was completely abolished upon treatment with H-89. SQ 22536, on the other hand, failed to reverse BML-111 induced VASP S-157 phosphorylation (Fig. 6c &d). Together, these findings suggest that BML-111 exerts inhibitory effects towards platelet by activation of PKA via a cAMP-independent route.
Fig. 6
BML-111 regulates VASP S-157 phosphorylation via cAMP-independent PKA activation. Human isolated platelets (4 × 108 cells/mL), under (a) Resting and (b) (0.1 U/ml) thrombin were pre-treated with BML-111 (12.5, 25 and 50 µM) or a vehicle-control (modified-Tyrode’s HEPES buffer) for 5 min then immunoblotted to detect VASP S-157 phosphorylation, a marker of PKA activity. Platelets were treated with PGI2 (1 µg/ml) as a positive control to stimulate the activity of PKA. Resting isolated human platelets (4 × 108 cells/mL) under resting conditions were treated with (c) H-89 (10 µM) or (d) SQ 22536 (100 µM) for 5 min before treatment with a vehicle-control (modified-Tyrode’s HEPES buffer) or BML-111(50 µM) for 5 min. Then, the samples were assayed for VASP S-157 phosphorylation. Treatment with PGI2 (1 µg/mL), which activates PKA by stimulating AC, was used as a positive control. Lysis of the samples was carried out using Laemmli sample buffer prior to separation by SDS-PAGE, then the samples were transferred to PVDF membranes. 14-3-3-ζ was detected by immunoblotting as a loading control. The impact of BML-111 on VASP S-157 phosphorylation is shown in representative blots from three distinct experiments. Data represent the mean ± SEM (n = 4). *P ≤ 0.05, ***P ≤ 0.001 and ****P ≤ 0.0001 were as calculated by One-way ANOVA
留言 (0)