Human samples
Human specimens were obtained from healthy individuals and individuals with AML registered at City of Hope National Medical Center, who consented to an Institutional Review Board (IRB)-approved protocol (IRB 14269). To isolate healthy peripheral blood mononuclear cells (PBMCs), fresh blood samples were processed using Ficoll-Paque Plus kit. To isolate leukemia stem cells (LSCs), bone marrow mononuclear cells (BMMCs) collected from R/R AML patients were stained with antibodies including Lineage (FITC-conjugated anti-CD2, anti-CD3, anti-CD4, anti-CD8, anti-CD14, anti-CD19, anti-CD20, anti-Mac-1, anti-CD56 and anti-CD235a), CD45 (BV510-conjugated anti-CD45), CD34 (PE-conjugated anti-CD34) and CD38 (BV605- or PE-Cy7-conjugated anti-CD38), and sorted through a BD FACSAria Fusion (BD Biosciences, San Jose, CA) for Lin−CD45dimCD34+CD38- population.
Animal study
The animal study was conducted under a protocol approved by the Institutional Animal Care and Use Committee at City of Hope (IACUC 22024). NOD/SCID/γ chain null (NSG) mice (female, 8–12 week old) were obtained from Jackson Laboratory (Sacramento, CA) and kept in micro-insulator cages in a pathogen-free condition. Mice were handled in laminar flow hoods. They were intravenously (i.v.) injected with 1 million AML blasts through tail veins. Five days after the injection, the mice were randomly divided into different treatment groups. 8CA (Tocris Bioscience, UK) (12.5 mg/kg/day, dissolved in PBS), 8AA (Santa Cruz Biotechnology, Dallas, TX) (5 mg/kg/day, dissolved in PBS) or vehicle control for 8CA/8AA was intraperitoneally (i.p.) administered. VEN (Selleckchem, Houston, TX) (20 mg/kg/day, dissolved in 10% ethanol, 60% Phosal 50 PG and 30% polyethylene glycol 400) or vehicle control for VEN was administered via oral gavage. The treatment lasted for 7 weeks. The bodyweight and temperature of the mice were measured once a week, and the survival was used as the endpoint measurement.
Cell culture
The cell lines MV4-11 and KG-1a were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and OCI-AML3 and Molm13 were obtained from DSMZ (Germany). They were maintained in Iscove’s Modified Dulbecco’s Medium (IMEM, Thermo Fisher Scientific, Irwindale, CA) containing 10% Fetal Bovine Serum (FBS, Thermo Fisher Scientific) and 100 U/mL penicillin/streptomycin at 37 °C with 5% CO2. Human cell lines purchased more than 6 months prior to submission of this manuscript and not frozen at an early passage were verified by ATCCs’ human short tandem repeat DNA profiling authentication service. The morphology of the cell lines was regularly monitored; potential mycoplasma contaminations were routinely monitored using a mycoplasma detection kit from Roche. All cell lines were mycoplasma-free.
Human LSCs were cultured using StemSpan serum-free expansion medium II (STEMCELL, Seattle, WA) supplemented with penicillin (100 U/mL) and streptomycin (100 mg/mL). Additionally, the medium was supplemented with stem cell factor (20 ng/mL), thrombopoietin (20 ng/mL), Flt3-L (20 ng/mL), IL-3 (10 ng/mL), and IL-6 (10 ng/mL).
Cell viability
Cells were seeded in 96-well plates at a density of 2000 cells per well and then treated with 8CA/8AA/VEN or their combinations at a series of concentrations for 48 h. MTT assay was used for colorimetric measurement of proliferation of AML cell lines, following the manufacturer’s instructions (Promega, Madison, WI). CellTiter-Glo assay was used to measure metabolic activity of primary AML cells following the manufacturer’s instructions (Promega).
Cell apoptosis
The Annexin-V and PI double-staining assay was used to determine cellular apoptosis. Briefly, cells were harvested and washed twice with Annexin-V binding buffer (BD Bioscience) and resuspended in 100 μL of the same buffer containing Annexin-V FITC and PI (BD Bioscience). The cells were then incubated in the dark at room temperature for 15 min, washed again and resuspended in 300 μL of buffer. The samples were then analyzed with an Accuri C6 flow cytometer (BD Bioscience).
Colony formation assay
Cells were counted after treatment and seeded onto 24-well plates using Human Methylcellulose Complete Media (R&D Systems, Minneapolis, MN) according to the provided instructions. The cells were then cultured for a duration of 10 to 20 days, adjusting the duration based on the density of colonies observed. Then the plates were scanned using an Observer 7 microscope (Zeiss, White Plains, NY) and the colonies were counted using ImageJ software.
AML patient dataset analysis
The Gene Data Set GDS1059 was obtained from NCBI GEO (Gene Expression Omnibus) and analyzed using the data analysis tools provided by NCBI Dataset Browser [22]. The dataset GS-DT-26 were acquired from GenomicScape (www.genomicscape.com) and analyzed using its survival analysis [23].
Flow cytometry
At room temperature and in the dark, LSCs were first stained with the LIVE/DEAD™ kit (Thermo Fisher Scientific) for 20 min. Afterward, the cells were fixed and permeabilized using a Fixation and Cell Permeabilization kit (Thermo Fisher Scientific). Next, LSCs were collected and incubated with the primary antibodies anti-MAT2A (Abcam, Waltham, MA) or anti-METTL16 (Abcam) for 1 h, and then washed and incubated with the secondary antibody Alexa Fluor® 647 (Abcam) for 30 min. Finally, LSCs were washed for flow cytometry analysis.
Peripheral blood samples from mice were collected and the red blood cells (RBC) within each sample were lysed using RBC lysis buffer (Santa Cruz Biotechnology). The cells were then washed and stained with APC-conjugated anti-human CD45 antibody for flow cytometry analysis.
Spleen samples from mice were collected and stained with a LIVE/DEAD™ kit (Thermo Fisher Scientific) for 20 min, and fixed and permeabilized using a Fixation and Cell Permeabilization kit (Thermo Fisher Scientific). Then the spleen cells were incubated with the primary antibodies anti-H3K4me3 (Cell Signaling Technology, Danvers, Massachusetts) for 1 h, and then washed and incubated with the secondary antibody Alexa Fluor® 647 (Abcam) for 30 min. Finally, the spleen cells were washed for flow cytometry analysis.
Flow cytometry analysis was performed using Fortessa X-20 flow cytometer (BD Biosciences), and the acquired data were analyzed using FlowJo V10 (Tree Star, Ashland, OR).
qRT-PCR
Total RNA was extracted from cells using Trizol (Invitrogen, Waltham, MA) and further purified with RNeasy kit (Qiagen, Germantown MD). Reverse transcriprion was performed using Omniscript RT Kit (Qiagen). Quantitative PCR reactions were run on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Irvine, CA) using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific). Primers were designed using Primer Bank and purchased from Integrated DNA Technologies. qRT-PCR analysis was conducted to measure the expression levels of the interested human genes (Supplementary Table 1).
Western blotting
Cells were lysed using M-PER™ Mammalian Protein Extraction Reagent containing Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). For histone extraction, the cells were lysed using Histone Extraction Kit (Abcam). The resulting products were mixed with loading buffer, heated at 95 °C for 5 min, and then cooled down to room temperature. The protein samples were then loaded onto SDS-PAGE gels (Bio-Rad) along with protein molecular weight markers (Bio-Rad) and electrophoresed at 100 V until the bands were separated sufficiently. The proteins were subsequently transferred to PVDF membranes using Trans-Blot Turbo Transfer System (Bio-Rad) for further analysis. The membranes were then blocked with blocking buffer (Li-Cor, Lincoln, NE) and incubated with primary antibodies at 4 °C overnight. Next, the membranes were washed with TBST and incubated with secondary antibodies at room temperature for 1 h, washed again and visualized using ChemiDoc Imaging System (Bio-Rad), or Odyssey Imager (Li-Cor).
RNA immunoprecipitation (RIP)
The RIP-qPCR assay was conducted as previously reported [24]. Briefly, cells were crosslinked with 1% formaldehyde for 10 min and then the reaction was stopped by 0.25 M glycine for 5 min. The cells were subsequently washed by PBS twice and lysed by RIP buffer supplemented with Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific), PMSF (Thermo Fisher Scientific), and RNase inhibitor (Invitrogen). After sonication for 10 cycles of 30 s on/30 s off at 4 °C using a Bioruptor Pico instrument (Diagenode, Denville, NJ), supernatant of cell lysate was harvested. 2 μg METTL16 antibody (IgG as control) was added to the supernatant. After incubation at 4 °C overnight, beads from Dynabeads Protein A Immunoprecipitation Kit (Invitrogen) were added and incubated for another 4 h. Beads were washed 3 times with RIP buffer. Finally, 100 µl elution buffer containing Proteinase K (Thermo Fisher Scientific) was used for further incubation at 42 °C for 1 h. Then the RNA can be extracted by Trizol or Phenol/Chloroform/Isoamyl Alcohol (Fisher BioReagents, Pittsburgh, PA) and then reverse transcribed to cDNA by using Omniscript RT Kit (Qiagen)according to manufacturer’s instructions. cDNA products with ~10-fold dilution were used as templates for qRT-PCR analysis.
Statistical analysis
Potential synergistic or additive effects of drugs were determined using CompuSyn software (Cambridge, UK). Synergism, addition, and antagonism effects were defined by combination index values of <1.0, 1.0, and >1.0, respectively.
Data are representative of at least three independent experiments, unless otherwise stated. GraphPad Prism 8.3.0 was used to statistically analyze data, and data were shown as Mean ± SD. A two-tailed unpaired t test was used to compare two groups under the same condition, and a two-tailed paired test was used to compare two groups under the same series of conditions. Ordinary one-way ANOVA mixed with Tukey’s multiple comparisons test were used to compare multiple groups. Log-rank (Mantel–Cox) test was used to compare the survival curves between two groups. P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.

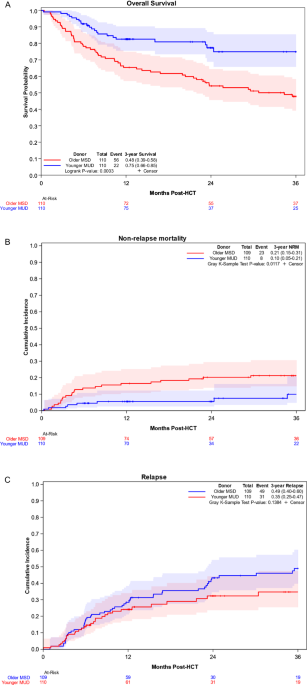


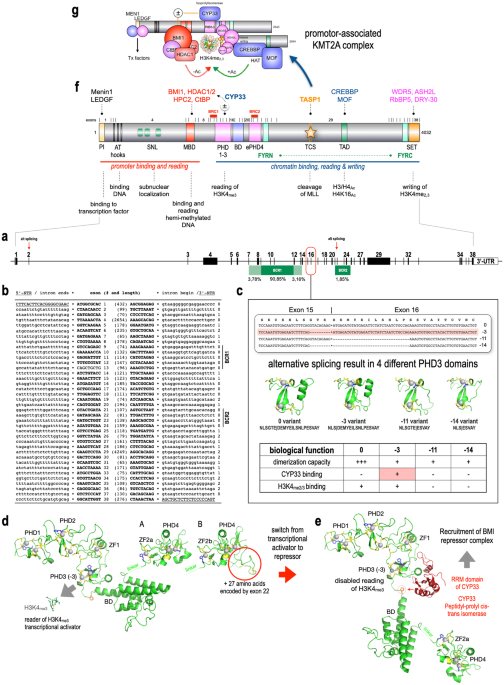
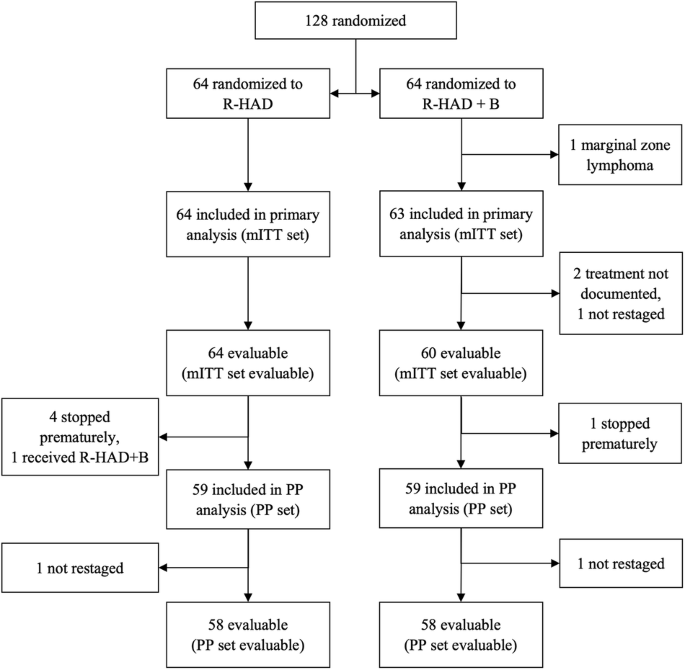
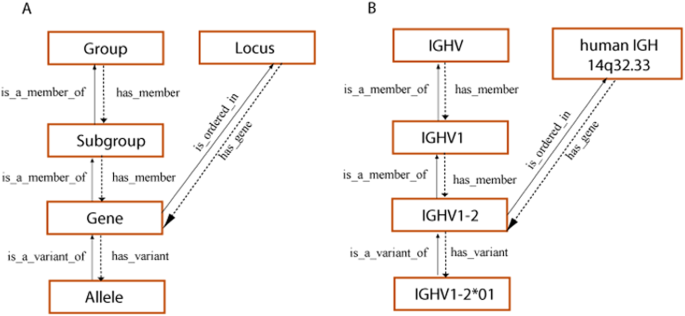

留言 (0)