
記住我









The average age of primiparous women has been gradually increasing since the 21st century, which is known to be negatively associated with reproductive outcomes (1–3). The advancement of assisted reproductive technology can compensate for the age-related decline in fertility, but evidence suggests that stopping or reversing the biological aging process is impossible (4, 5). Patients with certain genetic and autoimmune diseases, or women with excessive dieting, long-term radiation interference, or postoperative chemotherapy for cancer, may experience a premature decline in ovarian function (6), resulting in early-onset ovarian dysfunction, also called premature ovarian insufficiency (POI). Age-related infertility is mainly related to the decreased quantity and quality of oocyte that limit a woman’s ability to conceive. Studies using animal models revealed several cellular and genetic dysfunctions are causally related or correlated with aging. Although the mechanism of ovarian aging remains unclear, possible explanations for female ovarian aging are summarized in Figure 1.
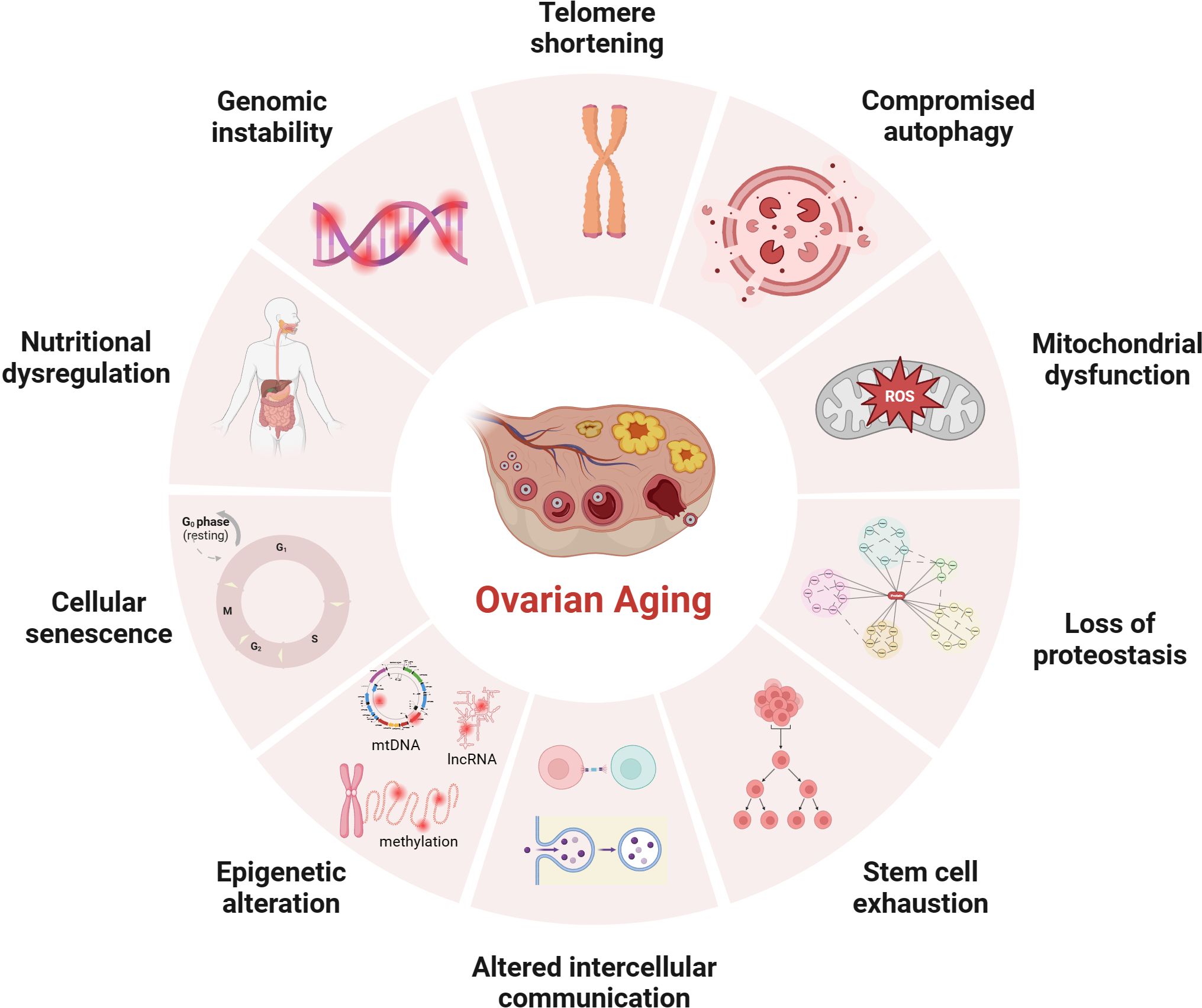
Figure 1 Potential mechanisms of ovarian aging (the figure was created with BioRender.com) Telomere shortening: Once telomere length becomes critically short, cellular senescence and apoptosis may occur during ovarian aging in mammals (7, 8); Compromised autophagy: The decrease of autophagic activity with age, likely leads to the accumulation of damaged macromolecules and organelles (9, 10); Mitochondial dysfunction: Mitochondial factors are explained further under subheadings in the text; Loss of proteostasis: Loss of proteostasis may damage the stability of microtubules and the integrity of meiosis in naturally aged mice oocyte (11); Stem cell exhaustion: Due to germ-line stem cells do not exist post-natally in female mammals (12–14), the reproductive potential of the ovaries will continue to decline after birth; Altered intercellular communication: There is an Increase In preantral follicles atresia in mice with suppression of intercellular junctions, which may be the cause of premature ovarian failure (15, 16); Epigenetic alteration: Studies have revealed the occurrence of epigenetic changes in the cells within the ovaries as age increases, including abnormal DNA methylation (17), histone modifications (18), and non-coding RNA-regulated modifications (19, 20); Cellular senescence: With the dysfunction of senescent cell, limited mitochondrial energy production, increased apoptosis of ovarian granulosa cells (21), increased inflammatory cytokines secretion (22) etc., would alter the microenvironment of follicle growth and affecting the quality and quantity of oocyte; Nutritional dysregulation: Dietary factors (23), low energy availability and high fat diet can increase the risk of POI; Genomic instability: In aging oocyte deceased efficacy of DNA repair mechanisms could cause low potentiation of anti-oxidant buffering and promote cell death (24).
Mitochondrial dysfunction provides a clue for explaining ovarian aging. Mitochondria is a kind of organelle with a double membrane structure and a diameter of 0.5~1.0 μm, containing limited Genetic material. Mitochondria is controlled by the mitochondrial and nuclear genomes. Human mitochondrial DNA (mtDNA) is a circular DNA molecule containing 16569 base pairs. All 13 proteins encoded by mtDNA are structural components of the electron transport chain (ETC), which assemble with nuclear-encoded proteins and become crucial components of the oxidative phosphorylation(OXPHOS) process. Mitochondria is the sites for oxidative metabolism in eukaryotes, and is the sites where sugars, fats, and amino acids ultimately oxidize and release energy. The common pathway responsible for the final oxidation of mitochondria is the tricarboxylic acid (TCA) cycle and OXPHOS, which correspond to the second and third stages of aerobic respiration, respectively. The glycolysis completed in the cytoplasmic matrix and the TCA cycle completed in the mitochondrial matrix produce high-energy molecules such as reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2). The role of OXPHOS in this step is to use these substances to reduce oxygen and release energy to synthesize adenosine triphosphate (ATP).
A big difference from somatic cells is that the number of mitochondria and the copy number of mtDNA in human mature oocyte are very high (6). These mitochondria are produced during the oogenesis stage, and the mitochondrial replication is believed to remain quiescent until the embryo reaches the blastocyst stage (25, 26). Using transcriptome sequencing, differential expression of mitochondrial-related genes was found by comparing the oocyte of young women and women with advanced age (27–30). It is reported that the increase in maternal age significantly decreased the number and quality of mitochondria in aging oocyte (31, 32). After ovarian aging, the morphology of mitochondria changed significantly, showing an increase in the number of swollen and vacuolated mitochondria (33–37). But ovarian functions can be partially restored by supplementing healthy mitochondria or improving mitochondrial function (38, 39), such as reducing the risk of mitochondrial oxidative damage caused by free radicals, reducing the expression of mitochondrial apoptosis-related proteins (40), and stimulating mitochondrial autophagy enhancers (41). The current manuscript reviews and summarizes the mechanism of mitochondrial damage in ovarian aging to explore possible intervention measures for improving and protecting the fertility of women with advanced age of childbearing age.
2 Mitochondrial dysfunction leads to ovarian aging2.1 The key role of mitochondrial oxidative stress damage in ovarian agingThe disruption of oxidative and antioxidant balance leads to excessive oxidative stress(OS), resulting in irreversible damage of ovarian. Reactive oxygen species (ROS) are byproducts resulting from energy production in the mitochondrial electron transfer chain to generate ATP. When the human body ages or is subjected to various harmful stimuli, excessive production of ROS and reactive nitrogen species (RNS) can disrupt the antioxidant defense barrier, trigger cellular OS response. The emergence of mitochondrial OS may be a mechanism leading to infertility, which is not unfamiliar to us. The high OS level detected in the human ovary is associated with follicular atresia, low fertilization potential of oocyte, the risk of aneuploidy, and sub-fertility (42, 43). Numerous studies have shown that during ovarian aging, the antioxidant enzymes in granulosa cells(GCs), and follicular fluid, such as superoxide dismutase (SOD), catalase (CAT), and Glutathione peroxidase (GSH-Px), were significantly reduced (44, 45). OS reaction is regarded as a specific initiator for oocyte aging (46), directly attacking the mitochondrial membrane and destroying the mitochondrial OXPHOS, resulting in reduced ATP production. Excessive ROS production directly affecting the target of the signaling pathway and acting as a second messenger interacting with intermediate reaction steps (47). ROS reduces the expression of isocitrate dehydrogenase 1 (IDH1) by activating the mitogen-activated protein kinase (MAPK) signaling pathway (48), activates the p53-SIAH1-TRF2 axis to induce telomere shortening (49), and promotes the aging of GCs. In addition to aging, GCs apoptosis is related to follicular atresia, which is an important mechanism of ovarian aging. Research has found that the strong oxidant H2O2 passes through HIF-1α signaling pathway to stimulates apoptosis of GCs (50). Peroxiredoxin 2(PRX2)deficiency accelerated age-related ovarian failure through the ROS-mediated JNK pathway in mice, and increased numbers of apoptotic cells (51). Another study showed that Ginsenoside Rb1 inhibits age-related GCs oxidative damage by activating Akt phosphorylation at Ser473 and by further interaction with FOXO1 (44). Therefore, the increase of OS plays an important role in the development of ovarian aging. The use of antioxidants to minimize OS may affect improving ovarian function women with advanced age (41, 43).
It is worth noting that when OS occurs, mitochondria will respond. Nuclear respiratory factors (NRF) can sense OS in the mitochondrial matrix and be activated. NRF1/2 can regulate the formation of mitochondrial respiratory chain complexes, transcription and replication of mtDNA. Loss of full-length NRF1 led to a dramatic increase in ROS and oxidative damages (52). In addition, SIRT1/3 also acts as a sensor by activating PGC1α- NRF1 promotes mitochondrial synthesis and coordinates cellular defense against ROS invasion (53, 54). Unfortunately, current research has found that NRF1, SIRT1, and SIRT3 are all downregulated in the ovaries of reproductive aging (55–57), indicating a decrease in the ability of mtDNA transcription and replication (mitochondrial synthesis). The antagonist response orchestrated by SIRT1 in oocyte seems to decrease with aging (53). When ROS accumulation exceeds the critical point, OS will further induce cell aging or apoptosis, affecting oocyte quality.
In summary, the increase of OS plays an important role in the development of ovarian aging by disrupting OXPHOS, inducing telomere shortening, and stimulating cell apoptosis (46–48). At the same time, the expression of molecules that receive ROS signals to activate mitochondrial synthesis is downregulated in the aging ovaries (55–57). The insufficient transcription and replication ability(mitochondrial synthesis) of mtDNA leads to a decreased ability to resist ROS in the cells.
2.2 Changes in mitochondrial genome during ovarian aging2.2.1 The decrease in mtDNA quantity leads to a decrease in the quality of aging oocyteWhen OS occurs within cells, mtDNA transcription and replication are driven to synthesize new mitochondria. Unfortunately, it has been found that mtDNA copy number decreases with ovarian aging (58, 59). The mtDNA is compacted into dense mitochondrial nucleoids. The mitochondrial transcription factor A (TFAM) binds to mtDNA and regulates mtDNA transcription activation in vivo by recruiting mitochondrial RNA polymerase (POLRMT) and mitochondrial transcription factor B (TFBM) for mtDNA replication (58). Current research has found that female individuals with TFAM recessive missense mutation showed the clinical phenotype of POI (59). Evidence up to date indicated the decreasing pattern of mitochondrial quantity and mtDNA content in mammalian oocyte with aging (31, 60, 61). In addition, a significant decrease in the copy number of mtDNA was found in unfertilized and degenerated oocyte of women with advanced age, with a notable positive correlation existing between the cytoplasmic volume of blastomeres from embryos and the mtDNA copy number (35). The decrease in mtDNA copy number is consistent with the difficulty in conceiving in patients with reproductive aging. The mtDNA copy number is an indicator of oocyte maturation and fertilization potential (62). The optimal mtDNA copy number and sufficient ATP level (at least 2pMol) are prerequisites for normal follicular development and maturation to ensure the good developmental potential of the fertilized blastocyst (63, 64).
On the other hand, the quality of oocyte is regulated by GCs (65). GCs include two types: mural granulosa cells(MGCs) and cumulus cells (CCs). Current research has found that, women with moderate expression of TFAM in the cytoplasm of human follicular fluid GCs exhibit better results in IVF (66). The mtDNA content of CCs and oocyte in women with diminished ovarian reserve (DOR) significantly decreased, while the quality of oocyte was positively correlated with the expression of TFAM mRNA in CCs (60). The copy number of mtDNA in GCs in POI patients and low prognosis in patients classified by POSEIDON negatively correlate with age (67). Due to the crucial importance of maintaining stable mtDNA copy number for maintaining mitochondrial function and cell growth (68), the decrease in mtDNA copy number in GCs is closely related to the occurrence of excessive cell apoptosis (69). Excessive apoptosis of GCs can lead to follicular atresia and decreased oocyte quality.
In summary, a significant decrease in the copy number of mtDNA in oocyte and GCs was found in individuals with reproductive aging (35, 60, 67). The decrease in mtDNA copy number is associated with a decrease in oocyte fertilization potential. In this context, some researchers have carried out mitochondrial transplantation techniques with the aim of increasing the number of healthy mitochondria in the cells, resulting in improved oocyte quality and pregnancy rates (70, 71). However, due to the small sample size of the study, further expansion is still needed to confirm the findings. Due to the high number of mitochondria and copy number of mtDNA in mature human oocyte (6). The decrease in mtDNA copy number may only affect one aspect of oocyte quality, and mtDNA mutations cannot be ignored.
2.2.2 The significant increase in mtDNA instability leads to a decrease in the quality of aging oocyteThe “mitochondrial aging theory” hypothesizes that mtDNA undergoes sustained oxidative damage, leading to the accumulation of harmful mutations (72). ROS formation takes place in close proximity to mtDNA, which lacks the protective measures of histone complexes and efficient DNA repair mechanisms, in contrast to nuclear DNA. Consequently, this vulnerability results in a heightened mutation rate (73). In situations such as aging, obesity, or chronic inflammatory stimulation, ROS production escalates, which renders mtDNA more susceptible to mutations (74, 75). The instability of mtDNA is manifested in various means, including mutations, base oxidation modifications, single-strand breaks, and so on. It has been found that harmful mtDNA mutations gradually accumulate in aging human tissues (76, 77). In addition, multiple studies have demonstrated that mutations in mtDNA encoding genes may lead to mitochondrial dysfunction and play a positive role in the pathogenesis of POI (28–30).
However, a few studies found no correlation between mtDNA deletion, rearrangement, and mutations of human oocyte and maternal age (75, 78–80). In their studies, the mtDNA mutation rate of oocyte was as high as 28~50.5%, and that of CCs was as high as 66% (78–80). The heteroplasmy created by mtDNA mutations are common in oocyte. The mtDNA bottleneck genetic mechanism during generational transmission is predicted to filter out the mitochondria population with mtDNA mutations effectively (81, 82). The difference in representative phenotypes and lower recurrence risk among women carrying heterogeneous mtDNA mutations demonstrate the effectiveness of the bottleneck theory (83, 84). Additionally, the number of primordial follicles in female mammals is fixed at birth, and there is no regeneration or renewal of the follicular pool after birth. It is unclear whether the prolonged quiescent stage before the resumption of meiosis could be affected by a hypoxic environment to undergo mtDNA mutations. In addition, during the oocyte’s maturation, there is a surge in mtDNA quantity and redox reactions (6), due to excessive ROS can also lead to mtDNA mutations, making it difficult to evaluate the occurrence time of mtDNA mutations.
Besides, nuclear encoded mitochondrial Helicase Twinkle (TWNK) and mitochondrial DNA polymerase γ (POLG) are essential proteins for mtDNA proofreading and fidelity (85–87). In humans, individuals with TWNK mutations exhibit Perrault syndrome (ovarian hypoplasia) (88), individuals with POLG mutations exhibit premature aging (89), genome wide association studies(GWAS) has found that POLG is associated with female menopause (89), and individuals with DOR have decreased POLG mRNA expression in CCs (90). POLG mutant mice showed a decrease in the number of mitochondria, abnormal mitochondrial distribution (aggregation and clustering), lower NADH/NAD+ redox ratio and weaker energy production, and a corresponding decrease in fertility (91, 92). The above research highlights the insight that mutations in nuclear coding genes lead to ovarian aging by affecting the stability of mtDNA.
In summary, many studies on mitochondrial gene expression have shown a link between decreased mtDNA copy number, accumulated mtDNA mutations, and ovarian failure. Genes are internal factors that determine the normal operation of mitochondria. The instability of the mtDNA genome will lead to dysfunction of executing proteins within mitochondria, which in turn will lead to a decrease in mitochondrial function. In fact, there is an endogenous protective mechanism within cells that is responsible for monitoring mitochondrial mass to maintain mitochondrial balance. Will the negative effects caused by mtDNA instability be recognized and eliminated? Therefore, it is necessary to further elucidate the relationship between mitochondrial quality testing and ovarian aging.
2.3 Disorder of mitochondrial quality monitoring mechanism in ovarian aging2.3.1 Mutation of the key gene CLPP in mitochondrial unfolded protein response leads to depletion of ovarian reserveMitochondrial unfolded protein response (UPRmt), as a typical mitochondrial nuclear signal transduction process, promotes the high expression of nuclear-encoded mitochondrial stress proteins by transmitting mitochondrial damage signals to the nucleus (93, 94), helping to restore mitochondrial protein balance, thereby protecting electron transport chain complexes to avoid proteotoxicity. The maintenance of mitochondrial protein function, including correct folding, aggregation, and necessary degradation, requires the involvement of proteolytic enzymes and molecular chaperones. It is known that caseinolytic peptidase P(CLPP) is a highly conserved serine proteolytic enzyme and a key enzyme in UPRmt (95). Currently, it is reported that human CLPP gene mutation has a clinical phenotype of Perrault syndrome or POI (96, 97). In animal experiments, CLPP-/- mice showed auditory defects and complete infertility (98), and the ovarian reserve in mice showed an accelerated state of depletion with age (99, 100). The reason maybe that CLPP-/- mice activate the Sirolimus target (mTOR) pathway in vivo (99), affecting the degradation of aggregated or misfolded cytochrome oxidase subunit 5A(COX5A) (97), blocking UPRmt, thus affecting the content and activity of complex IV in ETC, accumulating ROS and reducing mitochondrial membrane potential (MMP), and finally activating the internal apoptosis pathway. It follows that mutations in the key gene CLPP, where UPRmt is affected, will lead to ovarian reserve depletion in mice and humans. Therefore, we hypothesize that ovarian aging may be associated with differential expression of the CLPP gene, but providing a definitive conclusion is challenging due to the paucity of studies on ovarian aging-associated UPRmt. The inhibition of UPRmt process means a decrease in the genes driving mtDNA replication and transcription in the nucleus, and a limitation in the synthesis of new mitochondrial proteins.
2.3.2 Obstacles in mitochondrial biogenesis in aging ovaryMitochondrial biogenesis is a strict regulatory process that activates signaling molecules such as NRF1/2, TFAM, and TFBM through peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC1α), driving the replication and transcription of mtDNA, translating it into proteins, and assembling it into new mitochondria (101). SIRT1/3 also activates PGC1α-NRF1 promotes mitochondrial synthesis (53, 54). When mitochondria sense the initiation of OS or UPRmt processes, mitochondrial biogenesis is activated to maintain mitochondrial balance. Current research found a significant downregulation of PGC1α expression in the ovaries of cyclophosphamide-induced injury in POI (54, 102). NRF1, SIRT1, and SIRT3 are all downregulated in the ovaries of reproductive aging (55–57). In the ovaries of SIRT3 gene knockout mice, there was a decrease in mitochondrial membrane potential, uneven distribution of mitochondria, and a decrease in mtDNA copy number (56). Ginsenoside Rg1 improves ovarian function by activating SIRT1 and coenzyme Q10 improves ovarian function by increasing the expression of SIRT1 in PGC1α in animal experiments (103, 104). In addition, Adenosine 5’-monophosphate-activated protein kinase (AMPK), which is associated with the onset of POI (105), can activate and interacts with PGC1α through various mechanisms (106). Research has found that exercise can increase intracellular energy metabolism, activate AMPK and PGC1α to increase intracellular calcium concentrations thereby increasing the induction of mitochondrial biogenesis through transcription factors (such as NRFs, TFAM) (101). Inhibition of the AMPK/SIRT3 pathway will induce mitochondrial protein hyperacetylation and mitochondrial dysfunction in pig oocyte (107). In summary, mitochondrial biogenesis is a regeneration process that maintains the number of mitochondria, replacing old and damaged mitochondria with new and healthy mitochondria. Mitochondria with decreased membrane potential will fuse with newly formed mitochondria (one aspect of mitochondrial dynamics), sharing an internal system to restore mitochondrial quality once again. However, the downregulation of key genes involved in mitochondrial biogenesis in aging ovaries may lead to obstacles in the regeneration process (55–57, 60).
2.3.3 Defects in mitochondrial dynamics of aging oocyteMitochondria undergo continuous transformation through fusion and division states, which are manifested by morphological remodeling of the mitochondrial cristae and fracturing and lengthening of the tubular network, in order to achieve physiological functions. Mitochondrial fusion protein (MFN) mediate the assembly and fusion of mitochondrial inner and outer membranes to form a homogeneous mitochondrial network to maintain quality. Optic Atrophy 1(OPA1) is a GTPase that promotes fusion of the inner mitochondrial membrane and maintains cristae integrity. Dynamin-related protein 1 (DRP1), as core protein in mitochondrial division, can promote rapid changes in mitochondrial division activity according to cellular needs. Existing studies found that, the absence of MFN1 and MFN2 in mice oocyte resulted in infertility phenotype and loss of follicular reserve (108, 109), and the expression of DRP1 in aging oocyte was reduced (110). Low expression of MFN2 is associated with mitochondrial damage and apoptosis in ovarian tissue in POI model mice (111, 112). Since activated JNK phosphorylates MFN2, causing it to be degraded via the ubiquitin-proteasome system, the expression of MFN2 can be restored after supplementation with hydrogen sulfide or glutathione, antioxidants, or JNK inhibitors, and the improvement of mitochondrial morphology contributes to cell proliferation in ovarian cancer (113). In goat ovarian granulosa cell, Neuromedin S(NMS) treatment upregulates the expression of OPA1, MFN1, and MFN2 in the presence of NMUR2 knockdown, maintains mitochondrial fusion capacity and function, and thus regulates steroidogenesis (114). Therefore, we speculate that there are mitochondrial motility defects in aging oocyte. Due to the ability of mitochondrial fission to separate damaged mitochondria with low membrane potential from the entire mitochondrial network. Damaged mitochondria either fuse with newly formed mitochondria or are cleared through mitochondrial autophagy. The downregulation of mitochondrial fusion and fission ability implies an inevitable decrease in mitochondrial quality.
2.3.4 Downregulation of mitochondrial autophagy in aging ovaryWhen mitochondrial damage cannot be repaired through fission/fusion, the role of mitochondrial autophagy becomes particularly important. Mitochondrial autophagy is achieved through the phagocytosis of damaged mitochondria by autophagosomes, primarily via the PINK1-Parkin pathway, which removes damaged and membrane potential loss mitochondria, and receptor-dependent mitochondrial autophagy such as BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL2/E1B 19kDa interacting protein 3-like (BLIP3L or NIX), or FUN14 domain-containing protein 1(FUNDC1) (115). Studies have shown that maintaining mitochondrial autophagy is crucial for prolonging the reproductive capacity and oocyte quality of C. elegans and mice (116, 117). In particular, mitophagosome formation defects and accumulated damaged mitochondria were observed in germinal vesicle (GV) oocyte collected from the ovaries of older mice (118), which may be caused by PINK1 and Parkin proteins age-related accumulation and the degradation of ras-related protein rab-7a(RAB7) ubiquitination modification (118, 119). RAB7 is a monomeric guanosine triphosphate(GTP)-binding protein and an essential regulatory factor for the late endosomal/lysosomal network (120). Following treatment with RAB7 activators, the fertility of mice was improved. Therefore, Jin et al. suggested that excessive ubiquitination of RAB7 inhibits mitochondrial autophagy that should have been activated during ovarian aging (118). Another research reported a therapeutic strategy to ameliorate oocyte quality and reproductive outcome by enhancing mitophagy in aged mice (117). CoQ10 significantly increased PINK1 and Parkin proteins and improved embryonic development in postovulatory oocyte of pigs (104). From the above findings, it can be surmised that mitochondrial autophagy is down-regulated in senescent oocyte. Defects in the formation of mitochondrial phagosomes lead to the accumulation of damaged mitochondria and a decrease in mitochondrial quality. The decline in mitochondrial function ultimately leads to ovarian aging. Although the above results are exciting. It should be noted that so far, most of our understanding of mitochondrial autophagy has been through detecting the expression of proteins related to related pathways. However, for some reasons, such as the instantaneous occurrence of mitochondrial autophagy leading to cell apoptosis, it is challenging to measure mitochondrial autophagy accurately (121, 122).
In summary, we have elucidated four aspects of mitochondrial quality monitoring that may be related to ovarian aging (as shown in Figure 2). Current researches indicate that mutations in the key gene CLPP of UPRmt can lead to depletion of ovarian reserves in both mice and humans. The expression of key genes involved in mitochondrial biogenesis, mitochondrial dynamics, and mitochondrial autophagy in aging ovaries has been downregulated. Therefore, various functions of mitochondrial quality monitoring in aging ovaries may have been downregulated to varying degrees. A decrease in mitochondrial quality usually means a decrease in mitochondrial cristae area and enzymes related to aerobic respiration in the matrix, resulting in insufficient mitochondrial energy supply and affecting the function of oocyte.
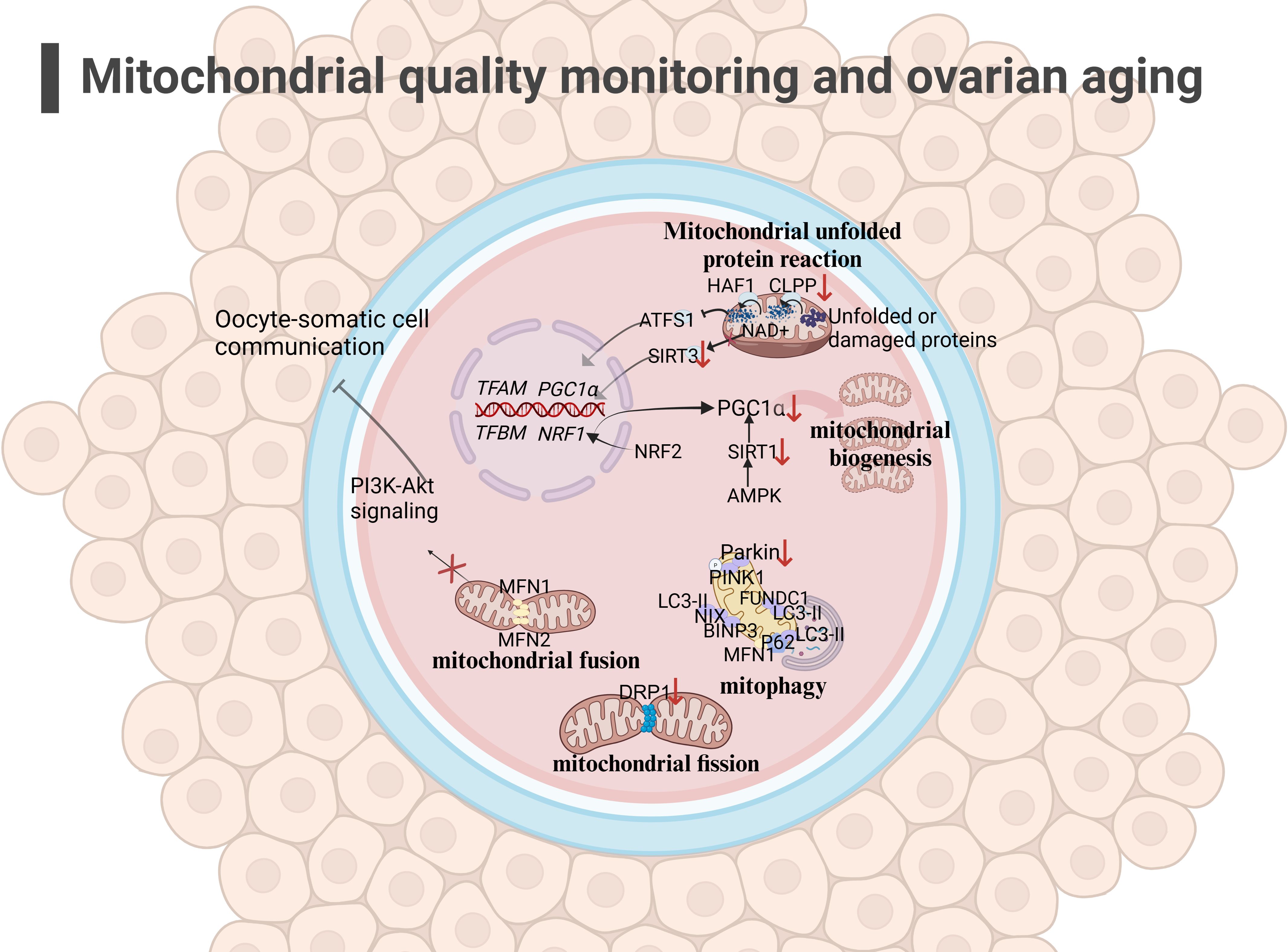
Figure 2 Mitochondrial quality monitoring and ovarian aging(the figure was created with BioRender.com) (1)mitochondrial fusion: The low expression of MFN1 ang MFN2 in the outer membrane in aging oocyte may affect mitochondrial fusion and inhibit the PI3K-AKT pathway, affecting oocyte-somatic cell communication (108, 109); (2) mitochondrial fission: The decreased expression of DRP1 in oocyte may affect mitochondrial function, leading to a decrease in oocyte quality (110); (3) mitophagy: Mitochondrial autophagy function decreases during ovarian aging (117, 118); (4) UPRmt: The decreased expression of CLPP and PGC1α in aging oocyte may hinder the occurrence of UPRmt (96, 97).
2.4 Mitochondrial dysfunction disrupts normal physiological function of oocyte2.4.1 Inadequate mitochondrial energy supply is a factor in meiotic errorsSome scholars have found that euploid oocyte screened from women with advanced age have implantation potential similar to that of young women with in vitro fertilization (IVF) (123). The production of aneuploid oocyte is the main reason for the decline in female fertility. The error of MI of female oocyte is considered to be the primary cause of human abortion and congenital defects (124, 125), and oocyte meiotic spindle morphology is a predictive marker of blastocyst ploidy (126), although mechanism of meiosis is complex. During meiosis, mitochondrial dysfunction significantly affects spindle assembly and ratio of kinetochore-microtubule connection (127, 128), and maternal age-related meiotic errors can be attenuated by reducing mitochondrial function (128). In the process of meiosis of mice oocyte, mitochondrial calcium uniporter protein (MCU) mediates the rapid entry of Ca2+ into mitochondria and provides high energy in an instant (129). The specific deletion of MCU leads to a low concentration of Ca2+ in mitochondria, low ATP levels, abnormal spindle assembly, and altered meiosis progression (129). Interestingly, the decrease of mitochondrial ATP concentration activates AMPK signal transduction. However, over-activated AMPK and Ca2+ overload result in OS, apoptosis, and meiotic cell cycle arrest and apoptosis in mammalian oocyte (130, 131). Above research demonstrated that precise mitochondrial Ca2+ homeostasis mediated by MCU is critical to oocyte meiosis. Moreover, the deletion of spindle defective protein 3 (SPD3) located on the outer membrane of mitochondria directly leads to the abnormal pairing of homologous chromosomes in the meiosis of Caenorhabditis elegans, which may be caused by the inhibition of mitochondrial function (132). Other proteins that regulate mitochondrial function, such as multidrug resistance protein 1 (MDR1) (133), regulate ROS efflux from the inner mitochondrial membrane, and RAB7 specific deletion regulates DRP1 phosphorylation (134), resulting in mitochondrial dysfunction, abnormal spindle shape, and increase of oocyte meiosis chromosome errors. In addition, SIRT3-/- mice aging oocyte exhibited spindle assembly interruption (55). These studies together highlighted the importance of mitochondrial energy supply for meiosis (as shown in Figure 3). In aging ovaries, the decline in mtDNA number and mutations, the down-regulation of mitochondrial quality monitoring mechanisms, and insufficient supply of mitochondria may persist, culminating in derangement of spindle assembly and motility, leading to an increase in meiotic chromosome errors in oocyte.
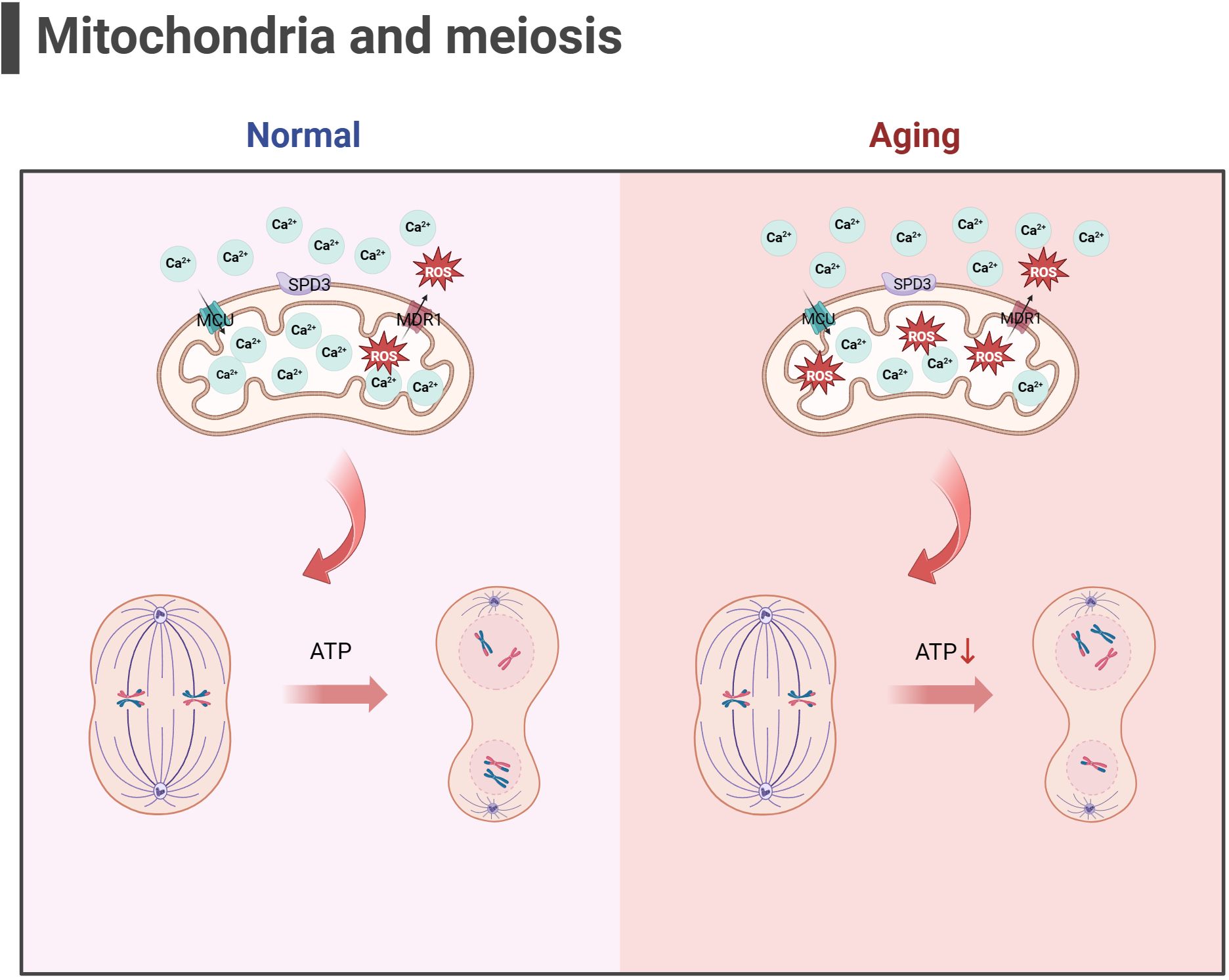
Figure 3 Mitochondria and meiosis in the oocyte (the figure was created with BioRender.com) MCU mediates the rapid entry of Ca2+ into mitochondria and provides high energy in an instant (129). MDR1 regulate ROS efflux from the inner mitochondrial membrane to maintain mitochondrial function (133). SPD3 may affect the abnormal pairing of homologous chromosomes which may be caused by the inhibition of mitochondrial function (132). Oxidative stress and mitochondrial ATP production disorders in aging oocyte may lead to abnormal spindle shape and increase chromosomal errors during meiosis in oocyte.
2.4.2 Mitochondrial dysfunction disrupts the oocyte-cumulus cell crosstalk in aging ovariesIn the cumulus oocyte complex (COC), CCs continuously consume glucose to supply metabolic intermediates (such as pyruvic acid) to oocyte, which will be crucial to energy metabolism in the mitochondria of oocyte (135). CCs are GCs surrounding oocyte that participate in the reproductive and maturation processes of oocyte through intercellular communication. MFN1-/- mice oocyte showed DOR phenotype, as well as damaged communication between oocyte and CCs (cadherin and connexin were downregulated), which may be caused by mitochondrial dysfunction and mitochondrial dynamics changes, the accumulation of ceramide in oocyte and the damage of PI3K-AKT signal transduction (109, 136). Ceramide is considered an important inducer of programmed cell death, and ceramide metabolic enzyme therapy can improve the quality of oocyte and embryos and the outcome of IVF (137). The PI3K-AKT pathway is an important signaling pathway for FSH to regulate glucose uptake in GCs and prevent ovarian aging (138). In addition, mitochondrial dysfunction of MII oocyte in AMPK -/- mice increased abnormally, PGC1α level decreased, ATP concentration decreased from normal, and connexin 37 and n-cadherin, which are involved in connection and communication between oocyte and CCs, were downregulated (139). The emergence of mitochondria-associated gene mutant mice with down-regulated expression of junctional gap junction proteins may have affected the delivery of some small molecules from the colobus cells to the oocyte, which in turn affected the quality of the oocyte. The above research provides evidence for mitochondrial dysfunction leading to abnormal communication between oocyte and CCs, supporting the theory of mitochondrial dysfunction leading to ovarian aging. However, there is a paucity of relevant studies, which need to be further clarified by rigorous experiments.
2.4.3 Hyperactivation of the mitochondrial apoptotic pathway is present during ovarian agingIn addition to energy generation, the important role of mitochondria in cells is also reflected in regulating cell apoptosis. Mitochondria is the central organelle of the apoptosis pathway, having an irreplaceable role in apoptosis. Under physiological conditions, Cytochrome c (Cytc) is the carrier for transferring electrons in ETC, establishing the mitochondrial transmembrane potential and generating ATP. BCL2 and BCL-xL form heterodimers with BCL2-associated X(BAX) and BCL2 homologous antagonist/killer (BAK), maintaining the integrity of the mitochondrial outer membrane and preventing mitochondrial apoptosis response. When stimulated by apoptosis, BAX/BAK forms an oligomer complex and inserts into the outer membrane pores of mitochondria, leading to changes in mitochondrial osmotic pressure, abnormal activation of Cytc release channels, and initiation of downstream caspase cascade reaction to induce apoptosis. Consistent with this, many studies have illustrated upregulated expression of apoptotic protein BAX, Cytc and Caspase 9 in the ovaries of mice with POF and POI animals (140–142), while the expression of anti-apoptotic protein BCL2 was downregulated. After intervention, the expression of these proteins was reversed, partially restoring ovarian function. Gene knockdown experiments have shown that BAX mutations prolong the fertility of female mice and alleviate health issues related to aging (143, 144). In addition, mitochondrial dysfunction and reduced ATP production may disrupt the normal physiological functions of cells (145, 146), leading to cell apoptosis. In summary, mitochondrial dysfunction can disrupt cellular antioxidant capacity and disrupt the OXPHOS process, hindering ATP production. Mitochondria themselves can perceive a decrease in membrane potential, sense apoptotic signals, and initiate the mitochondrial apoptotic pathway. The excessive activation of mitochondrial apoptosis pathway is likely closely related to the occurrence of ovarian aging (as shown in Figure 4). Because there is excessive apoptosis of GCs and increased follicular atresia during ovarian aging.
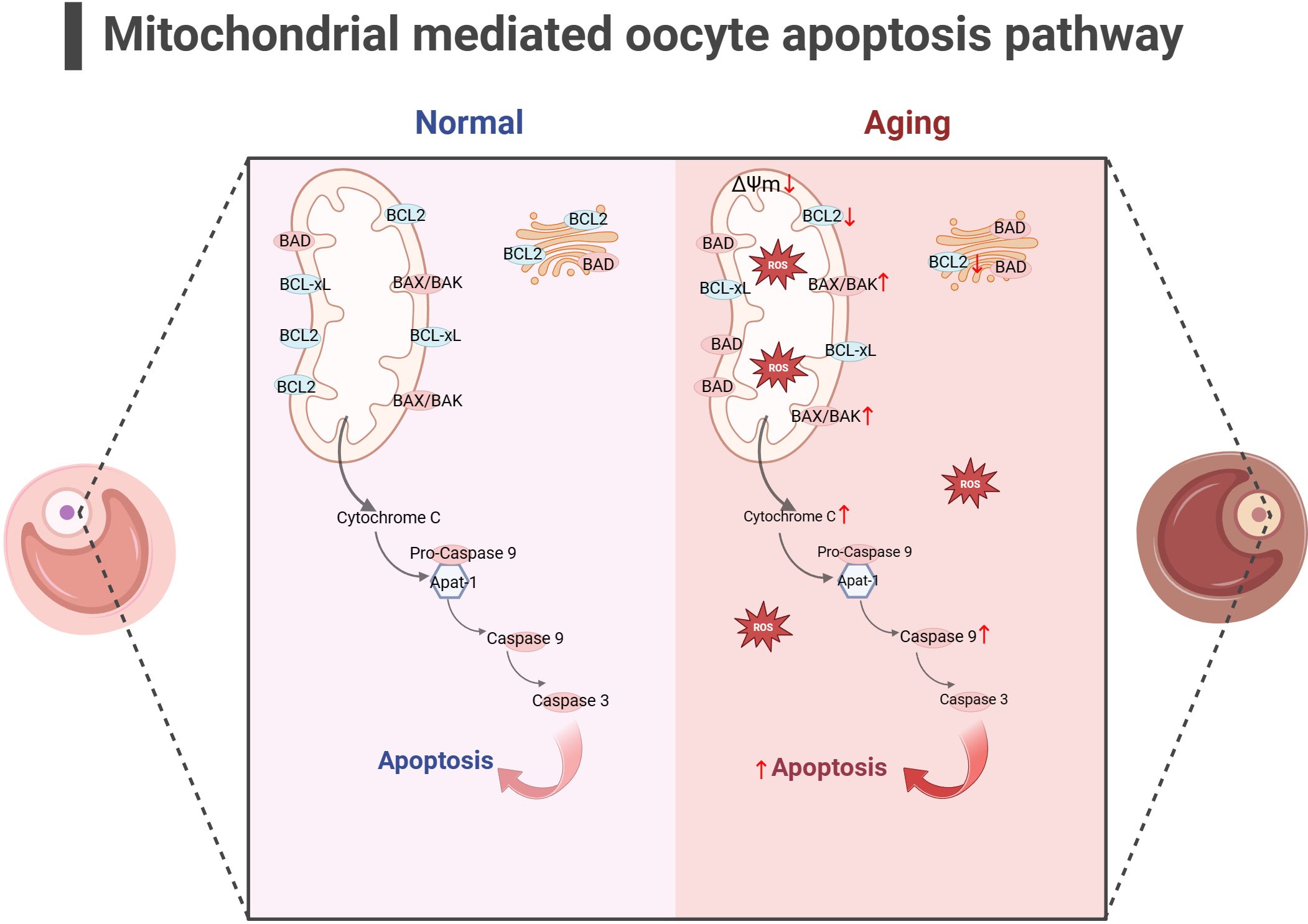
Figure 4 Mitochondrial-mediated oocyte apoptosis pathway (the figure was created with BioRender.com) In aging oocyte, excessive cell apoptosis signals are activated under stimuli such as mitochondrial oxidative stress and decreased mitochondrial membrane potential. At the same time, the expression of anti-apoptotic proteins such as BCL2 decreased, while the expression of apoptotic proteins such as BAK, CytC and Caspase9 increased (140–142).
2.4.4 The link between mitochondrial dysfunction and telomere damage as one of the latest mechanisms of ovarian agingIn addition, the telomere theory is one of the latest mechanisms to explain female reproductive aging (147).Telomeres are nucleoprotein complexes at the end of chromosomes, maintaining the integrity of chromosomes and inhibit DNA damage reactions at the free end of chromosomes (148). It is widely recognized that telomeres are susceptible to oxidative damage, and OS markers have been shown to exhibit a negative correlation with telomere length (149). Due to the significant features of ovarian aging, such as reduced ATP production and increased ROS accumulation (88), telomere loss and telomere-driven replicative senescence caused by OS may be important mechanisms of ovarian aging (150). In addition, there is a link between telomere function and mitochondrial biosynthesis. Existing studies have found decreased telomerase activity in ovarian GCs of POI patients (151, 152). The lack of telomerase activates p53 which in turn binds and represses PGC1α and PGC1β promoters, thereby inhibiting mitochondrial biogenesis (153, 154). The dysfunction of mitochondrial biosynthesis leads to the decline of cell resistance to OS and further accelerates the modification of 8-hydroxy deoxyguanosine on the telomere guanosine base. In addition, the link between telomere shortening and mitochondria is also mediated by NAD+-SIRT1-PGC1α axis establishment (124, 155, 156). The DNA repair process after telomere damage consumes NAD+, and the loss of SIRT1 activity will further affect the mitochondrial biogenesis mediated by PGC1α (155). In aging ovaries, the decrease in SIRT1 expression is a common phenomenon (55). Thus, the excessive occurrence of mitochondrial OS may accelerate telomere shortening at the ends of chromosomes, which in turn can inhibit the expression of the key gene for mitochondrial biosynthesis, PGC1α, through the activation of P53, leading to a decline in intracellular mitochondrial function, which further affects the DNA repair process. Ultimately, this vicious cycle of the above leads to the onset of ovarian senescence (as shown in Figure 5). Given the current research results, it is difficult to explain the sequence between telomere damage and mitochondrial dysfunction. Still, the two seem to interact with each other to lead to ovarian aging in women.
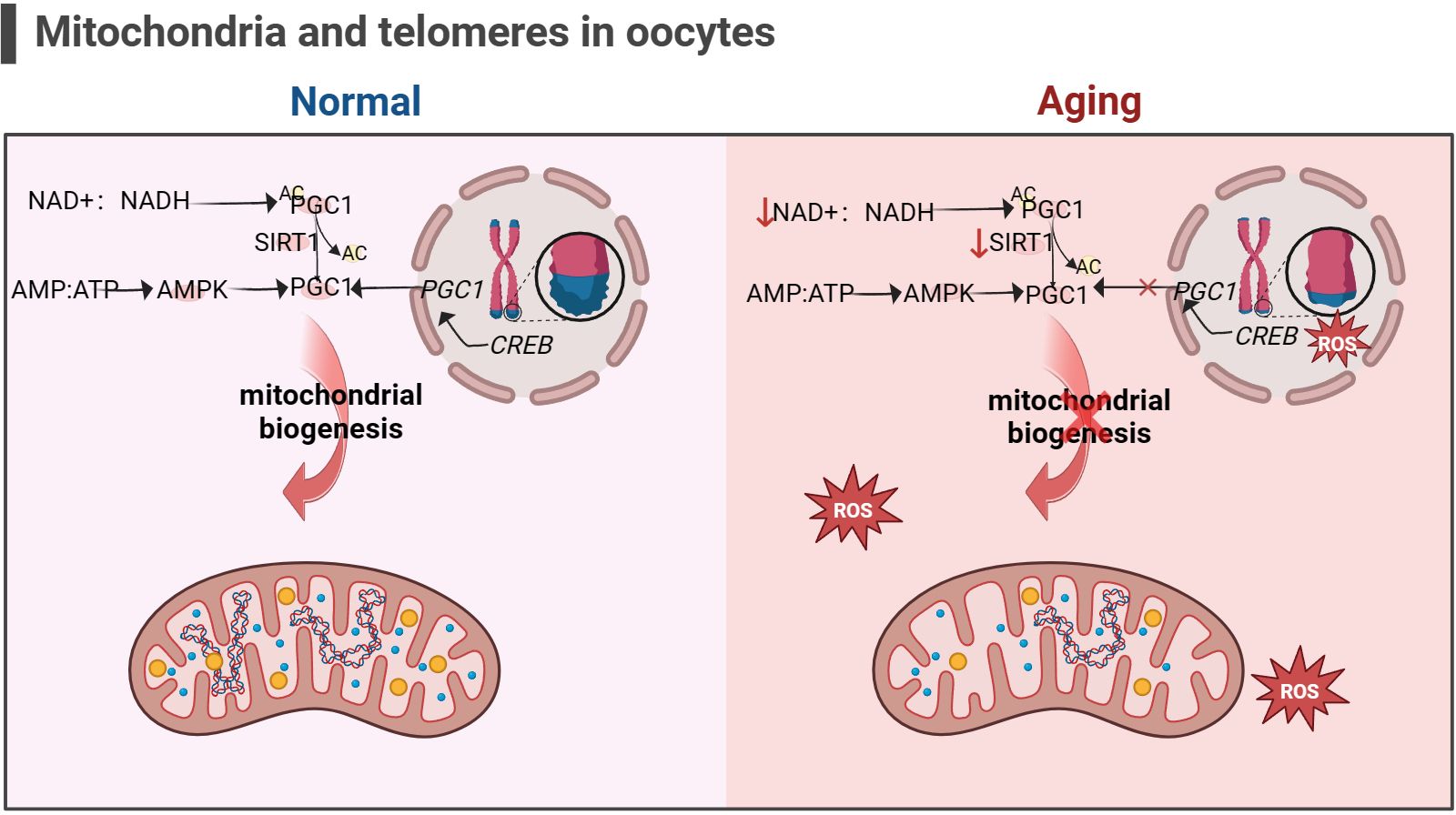
Figure 5 Mitochondria and telomeres in the oocyte (the figure was created with BioRender.com) The cAMP response element-binding protein (CREB) is a co-factor for PGC1 expression. Abnormal telomere function can activate p53, thereby inhibiting the expression of PGC1. The decrease in PGC1 expression in turn inhibits mitochondrial biogenesis, leading to a decrease in the ability of cells to resist oxidative stress (156). In addition, increasement in the proportion of intracellular AMP/ATP and NAD+/NADH also promotes PGC1 expression, but this is not suitable for aging cells, as the content of NAD+ and SIRT1 significantly decreases (53, 54).
2.4.5 Epigenetic regulation involving mitochondria influences oocyte senescenceOocyte quality decline during ovarian aging occurs in part through epigenetic regulation. Intermediates generated by metabolic processes within mitochondria can generation and modify nuclear epigenetic marks to achieve important mediators of mitochondrial-nuclear communication (157). Histone methyltransferases (HMTs) and histone demethylases (HDMs) are responsible for histone methylation status. S-adenosyl methionine (SAM) produced by the cytoplasmic methionine homocysteine cycle in the mitochondrial folate cycle is a donor of histone methyltransferases (HMTs) (158). Besides, the acetyl-CoA-producing enzyme ATP-citrate lyase (ACL) also regulates DNA methyltransferase 1 (DNMTl) (159), which affects the level of DNA methylation. Pyruvate, ketones, amino acids, citrate, acetate, and beta oxidation of lipids can produce acetyl-CoA, which is the substrate for the histone acetyltransferases (HAT) (160). Under conditions of energy and acetyl-CoA enrichment, this leads to histone acetylation and gene transcription. Histone demethylases (HDMs) comprise two major classes: lysine-specific demethylases (LSD) and Jumonji C domain demethylases (JMJD). LSD1 catalyzes the demethylation of mono- or dimethylated H3K4 and H3K9, whereas LSDs act as receptors in the mitochondrial ETC (161). JMJD-mediated demethylation of histones requires α-ketoglutarate (162), a substrate produced mainly by the tricarboxylic acid (TCA) cycle in the mitochondrial matrix. Current research has found that low methylation of DNMT1/DNMT3a/DNMT3b/DNMT3L promotes oocyte aging (17). Specific disruption of LSD1 led to a significant increase in autophagy through its H3K4me2 demethylase activity and a decrease in the number of oocyte in perinatal mice, leading to depletion of oocyte (163). Epigenetic enzymes recognize, add, and remove epigenetic markers on DNA and histones. Due to the association between the formation of epigenetic enzymes and mitochondrial metabolism, the transmission of mitochondrial metabolite levels or stress signals may lead to various epigenetic changes. Unfortunately, few studies have directly explored the association between mitochondrial epigenetic regulation and ovarian aging.
In summary, mitochondrial dysfunction during cellular aging may lead to spindle assembly and motility during meiosis by affecting energy supply. Both MFN1-/- and AMPK -/- mice showed mitochondrial dysfunction and abnormal communication between oocyte and GCs. In addition, the occurrence of mitochondrial dysfunction may directly initiate the mitochondrial apoptosis pathway, leading to follicular atresia. Excessive OS during aging not only activates the mitochondrial apoptosis pathway, but may also accelerate telomere shortening. Telomere shortening hinders the recovery of mitochondrial function by inhibiting mitochondrial biosynthesis. Finally, mitochondrial metabolites may also affect the expression of epigenetic enzymes and promote ovarian aging.
3 Methods to slow down ovarian aging and prolong reproductive lifespan by intervening in mitochondrial functionAt present, there is no effective technology in clinical practice to prevent the occurrence of female reproductive aging, and the current treatment methods are still in the exploratory stage. However, it is worth celebrating that some animal experiments have shown great potential in improving female reproductive function. Below we will discuss each of the currently discovered therapeutic mechanisms of drugs for mitochondrial damage.
3.1 Mitochondrial nutrition therapyWe summarize the therapeutic drugs targeting mitochondria to prevent ovarian aging in Table 1 and classify them according to mitochondrial energy metabolism, quality control, and mitochondrial apoptosis pathways.
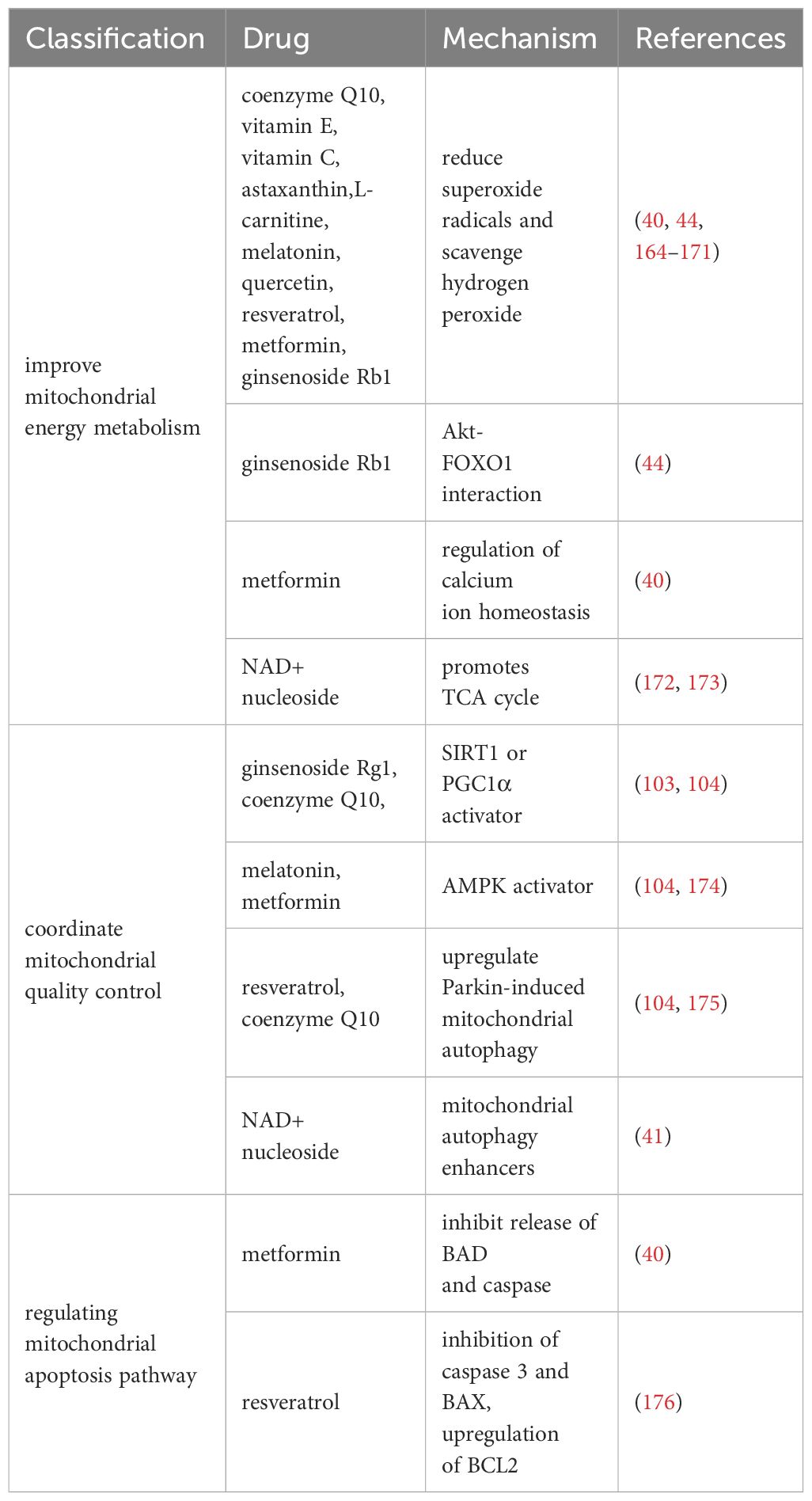
Table 1 Drugs that improve mitochondrial function.
3.1.1 Drugs for improving mitochondrial energy metabolismResearch has shown that multiple antioxidants can restore mitochondrial function by reducing ROS production and stimulating antioxidant production. It mainly includes coenzyme Q10 (177), vitamin E (164), vitamin C (164), L-carnitine (165, 166), melatonin (167, 168), quercetin (169), resveratrol (170), astaxanthin (171), and ginsenoside Rb1 (44).
In addition, ginsenoside Rb1 also promotes Akt binding to FOXO1 and inhibits OS occurrence (44). Metformin regulates calcium homeostasis to prevent follicular atresia of aging ovaries of Laying Chickens (40). NAD+/NADH, or coenzyme I, is a redox cofactor and enzyme substrate in mitochondria, critical for energy metabolism, DNA repair, and epigenetic regulation in cells. In the ovaries, the level of NAD+ decreases with age in a dependent manner (172, 173). After supplementing NAD+ precursors (NR or NMN), ovarian function in elderly mice was restored, and the mitochondrial TCA cycle was improved at the micro level (172, 173).
3.1.2 Drugs for coordinating mitochondrial quality controlNormal mitochondrial function and internal environmental homeostasis require close coordination between mitochondrial biogenesis and clearance. Compounds coordinating mitochondrial quality control are expected to become effective therapeutic interventions for ovarian aging. It is reported that ginsenoside Rg1 can increase SOD, CAT levels, and activate SIRT1 in POF mice model (103). CoQ10 significantly prevented aging-induced oxidative stress, and increased mitochondrial biogenesis (SIRT1 and PGC1α) and mitophagy (PINK1 and Parkin)-related proteins in postovulatory oocyte of pigs (104). Resveratrol can increase the expression of PINK1 and Parkin proteins regulate the autophagy ability of mitochondria, and protect ovarian function (175). Metformin and Melatonin are AMPK activators (105, 174), which could increase intracellular calcium concentrations thereby increasing the induction of mitochondrial biogenesis through transcription factors (such as NRFs, TFAM) (111). After 20 weeks of treatment with NMN in 40-week-old mice (41), mitochondrial biogenesis, autophagy level and protease activity in ovarian GCs were increased, and the ovarian reserve was rescued to some extent.
3.1.3 Drugs that regulate mitochondrial apoptosis pathwaysThe study found that Resveratrol treatment reduced the expression of mitochondrial apoptosis promoting protein caspase 3 and BAX and upregulated BCL2 expression in POI model mice (176). Metformin can inhibit the release of mitochondrial apoptosis factors (BAD and caspase) in Laying Chickens (40). However, whether these benefits observed in animal models can be replicated in humans remains to be determined.
Currently, various potential drugs targeting mitochondria have been developed (Table 1). However, these drugs are not necessarily suitable for preventing ovarian aging. Various drugs have been found to have positive effects in resisting OS, improving OXPHOS efficiency, promoting mitochondrial occurrence and autophagy, and inhibiting mitochondrial apoptosis in animal experiments. Whether these benefits observed in animal models can be replicated in the human body remains to be determined.
3.2 Mitochondrial replacement therapyMitochondrial transplantation was initially primarily used to prevent disease transmission caused by mtDNA mutations, becoming a viable alternative to avoiding damaged mitochondrial offspring inheritance. Subsequently, due to the increasingly important role of mitochondria in human reproduction, people began to think about improving the quality of gametes by enhancing the quality of mitochondria.
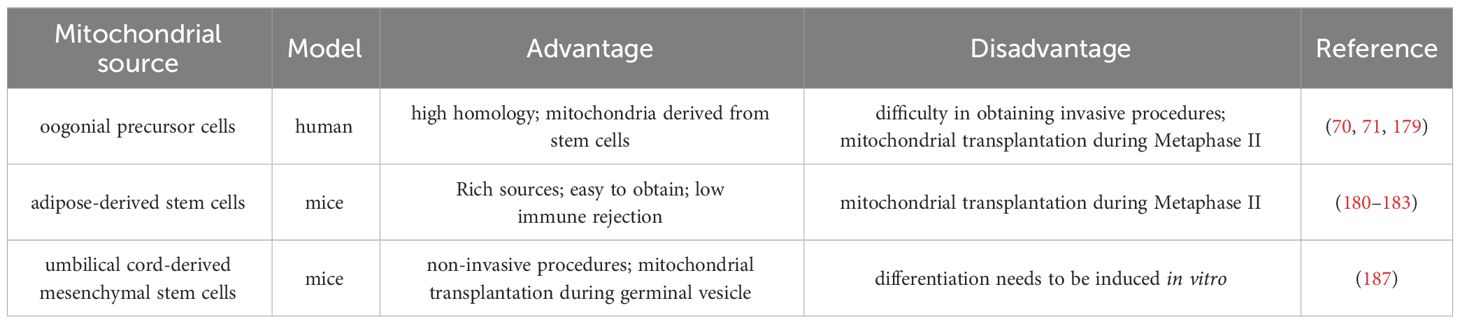
3.2.1 Autologous mitochondrial transplantation technologyThe autologous mitochondrial transplantation technology (178), focuses on increasing the number of healthy mitochondria in cells while avoiding the introduction of third-party DNA in gametes. It is reported that injection of mitochondrial from oogonial precursor cells(OPCs) while intracytoplasmic sperm injection (ICSI) can improve the quality of oocyte and pregnancy rate in the ICSI cycle of women with multiple IVF failure (70, 71). Unfortunately, a more rigorous tri-blinded, randomized, and single center-controlled study conducted in Spain in 2019 found that mitochondrial transfer of OPCs did not improve embryonic development potential and pregnancy rate in patients with previously IVF failed (179). Recently, it has been confirmed that autologous adipose stem cells (ASCs) mitochondrial transplantation can improve the quality of oocyte in juvenile or aged mice (180–182), while Sheng et al. found negative results (183). It should be noted that the mitochondrial dysfunction of aging oocyte seems to have occurred long ago. Most of the common aneuploid oocyte originate from MI (126, 184). It may be too late to save the quality of MII oocyte by ICSI injection of active mitochondria. Furthermore, the origin of mitochondria is an issue, and whether oogonial stem cells exist and how to obtain them is a highly controversial issue (185, 186). In summary, the efficacy of the above mitochondrial transplantation methods in improving ovarian function is controversial, and they are not considered standard treatments.
Tang et al. have established a noninvasive optimized autologous mitochondrial transplantation technique: inducing mice autologous umbilical cord mesenchymal stem cells into GCs (iGCs) and co-cultured them with weakened zona pellucida GV in growth differentiation factor 9 (GDF9) containing medium for three days. Tang et al. have found that mitochondria migrate from iGCs to GV oocyte through transzonal filopodia, significantly improving these oocyte’ maturation rate, quality, and developmental potential (187). The birth rate of aged mice after embryo transfer has also been improved (187). In Table 2, we summarized the mitochondrial transplantation methods of from different sources. Due to its non-invasive and early intervention characteristics, it is worth further research. In the future, we look forward to researching and solving the source of mitochondrial acquisition, achieving timely and non-invasive transplantation as much as possible, and providing promising strategies for improving the quality of oocyte and the fertility of women with advanced age.
留言 (0)