
記住我
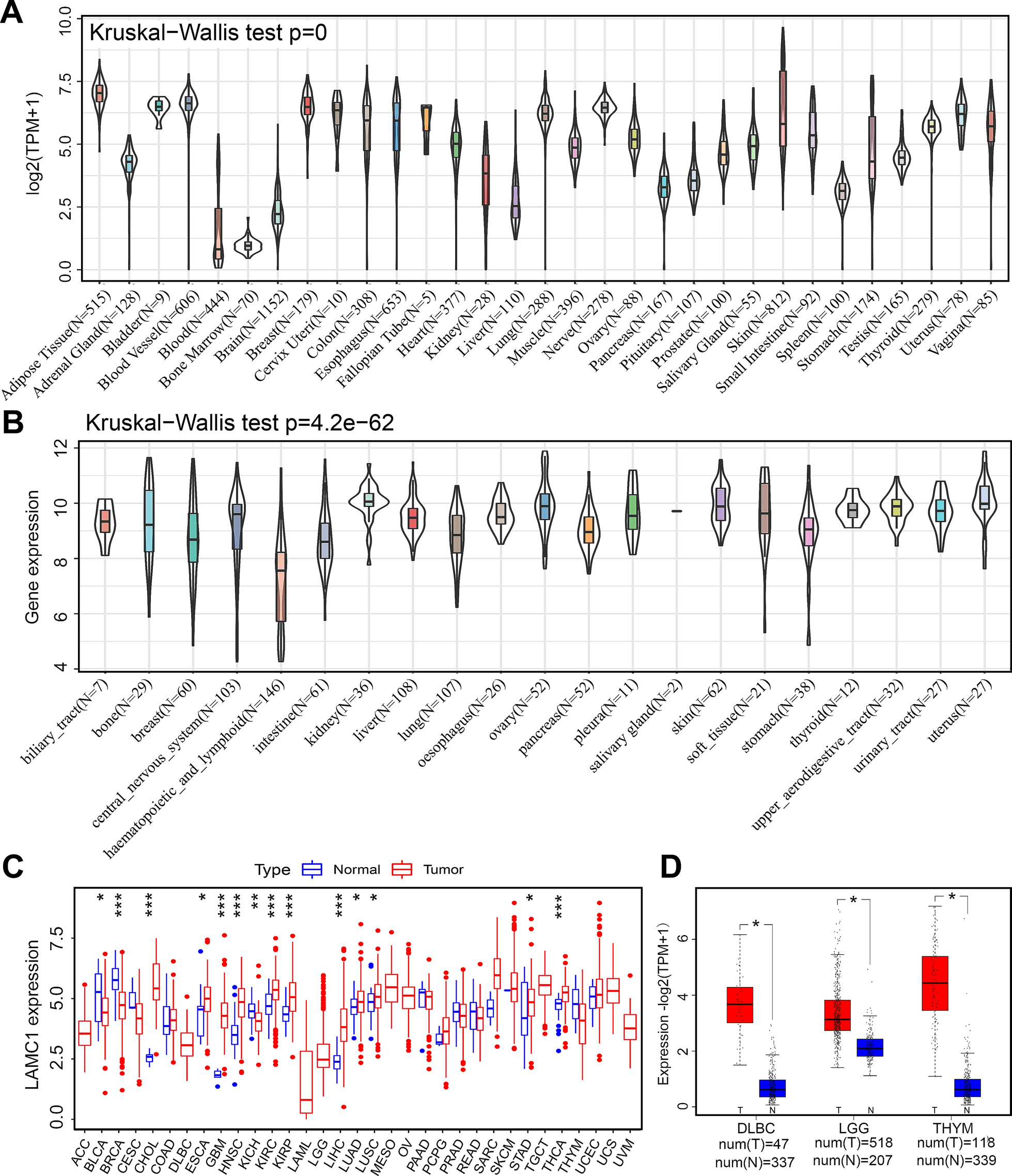




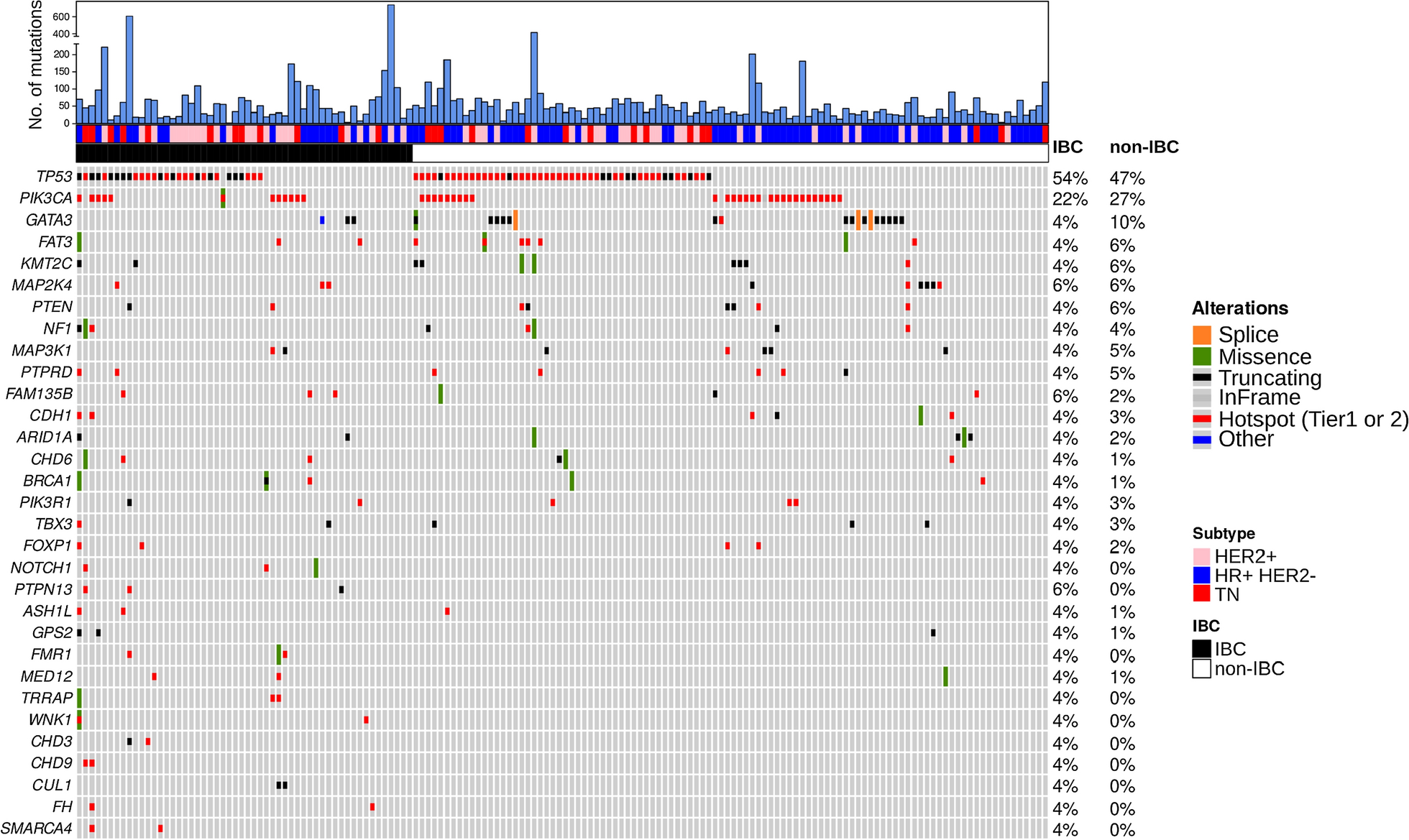
Here we describe a computational framework to perform machine learning and feature selection algorithms on large-scale genotyped data. Figure 1 provides a graphic overview of the main steps. The first step, depicted in Fig. 1A, aimed at gathering individuals for analyzing CAD phenotypes.
Fig. 1
Computational framework comparing different feature selection strategies for the selection of risk loci panels for CAD. A–C The data collected from the UKB is subjected to preprocessing to extract the phenotype of interest (CAD vs. non-CAD) and generate high-quality genotype imputed data. D, E To identify the most relevant covariates for association tests, we performed an analysis of CAD-associated risk factors and conducted principal component analysis on the genotype data. F tenfold cross-validation was used to perform a fair comparison between ML-based methods and PRSs. G Genomic variants for predicting CAD were selected based on three feature selection strategies encompassing filter-based and embedded methods. H, I Three PRS methods were implemented and combined with Logistic Regression-based classifier for the classification. J Genomic variants selected through FS were systematically uses to train three different classification algorithms: Lasso, RF and SVM. K The Area Under the ROC Curve (AUC) statistics as the main accuracy metric. Moreover, we also recorded the frequency of each feature being selected across different training set and feature selection methods. L The most informative SNPs were further analyzed to assess their biological relevance
We defined CAD and non-CAD phenotypes using ICD-9, ICD-10, and OPCS-4 codes. Refer to Additional file 1: Table S1 for a comprehensive list of diagnoses and health statuses considered associated with the CAD phenotype. Moreover, at this stage, kinship estimates are computed to identify related individuals, who are then removed before performing association tests, calculating PRS scores, and applying ML-based feature selection methods across the CV iterations. Imputation and quality control were performed to generate the initial set of single nucleotide polymorphisms (SNPs), which are genetic variations in a population at specific positions in the DNA sequence (Fig. 1B–C). Imputation refers to the process of filling in missing data in a dataset, while quality check refers to removing markers and individuals with low quality or that are unlikely to contribute to association analysis, thus increasing the power and accuracy of downstream analyses. Quality control on the genetic markers included checking for markers that were in linkage disequilibrium (LD), which can lead to redundant information, and markers that had high levels of missing data or low minor allele frequency (MAF). Markers that failed these quality control criteria were removed from the analysis. Association tests between CAD and known risk factors were performed and principal component analysis (PCA) was applied to genotype data to uncover population structure (Fig. 1D–E). The steps illustrated in Fig. 1F–K shows the machine learning framework which aims to compare PRS with standard ML and feature selection algorithms. In more details, three feature selection strategies, encompassing filter-based and embedded methods, were used in combination with three classification algorithms (Fig. 1G). These methods were then compared against three PRS strategies (Fig. 1H–I). Machine learning helps identify new risk loci for complex diseases, selecting genetic variants that, when combined, yield higher prediction accuracy compared to PRS-based approaches. To guarantee a fair comparison, PRS was defined within the training set and evaluated on an independent test set, preventing bias. The computational framework offers a more accurate assessment, enabling informed comparisons between ML and PRSs. 10-folds cross validation was used to more robustly estimates prediction performance of all tested methods (Fig. 1F), while the Area Under the ROC Curve (AUC) statistics was used to as main accuracy metric (Fig. 1K). Subsets of SNPs that are most informative for the identification of the CAD phenotype, which were selected through ML-driven feature selection algorithms, were further analyzed to assess their biological relevance, by using the tool Functional Mapping and Annotation (Fig. 1L). For further details on the methodology used in this study, please refer to the “Materials and Methods” section.
Population characteristics and association between CAD and known risk factorsWe examined the UKB population characteristics and associations between CAD patients and known risk factors across the entire UK Biobank population. Baseline characteristics of CAD are summarized in Table 1 by sex, age and commonly used risk factors. CAD participants’ average age is 60.33 years, and non-CAD controls’ average is 56.24 years. Triglycerides are slightly higher in CAD cases, as expected [22].
Table 1 Baseline characteristics of UK Biobank participants included in the present studyLDL and total cholesterol values are higher in non-CAD cases, likely due to statin treatment in CAD patients, which reduces both LDL and total cholesterol levels. Associations between known risk factors and CAD cases were then estimated through logistic regression analysis. The logistic regression model also included basic covariates (e.g., sex and age), ethnic background, genotype batch, assessment center, and PCs adjusting for population structure. Figure 2 shows odds ratios and 95% confidence intervals from multivariable ordinal regression, indicating the association between cardiovascular risk factors and CAD cases. HDL is a protective factor, while LDL and cholesterol show no increased risk, likely due to statin treatment in CAD-diagnosed individuals [23, 24]. Principal component analysis (PCA) was applied to genotype data to uncover population structure and use the PCs as covariates (Additional file 1: Fig. S2) for subsequent analyses.
Fig. 2
Odds ratios of traditional cardiovascular risk factors. A larger odds ratio indicates a stronger association between the risk factor and CAD. The red color is used to indicate significant associations (p < 0.05). The vertical line at x = 1 indicates an odds ratio of 1, in which case there is "no effect". For the calculation of odds ratios pertaining to traditional cardiovascular risk factors, we considered the complete set of UK Biobank individuals or non-CAD, which consisted of 467,215 and 35,290 individuals for the respective categories
Additional file 1: Fig. S3 includes the SHAP summary plot, which shows the contribution or the importance of each feature, including risk factors, batches, and PCs, on the CAD risk and their effect on the single predictions. Shapley values help detect risk factors, batches, or PCs affecting predictions. Features are ranked by their ability to improve predictions, with age and sex having mild contributions. Genotype batch and assessment center show no contributions to CAD status, while vascular heart problem diagnosis has a high positive contribution. However, it should be acknowledged that the lack of diagnosis is not necessarily associated with non-CAD cases. Additional file 1: Fig. S4 shows correlation and VIF analysis outcomes, assessing multicollinearity between predictor variables in a CAD status regression model. High VIF score variables were removed, specifically total cholesterol and ethnic background, represented by LDL/HDL levels and the first 20 PCs. The final set of covariates was used for genotype-CAD association analysis, generating summary statistics for CAD-relevant single-nucleotide variants. It should be noted that summary statistics are systematically computed within each generated training dataset in a cross-fold validation framework, as they are used in one of the feature selection strategies to select risk loci evaluated in this study and to build PRSs. This aligns with the machine learning framework, where feature selection and model training must be implemented within a training set and evaluated within an unseen test set to avoid data leakage [25].
Using PRS and ML methods for CAD prediction based on panels of genetic variantsPredicting phenotypes with panels of genetic variants captures cumulative effects, reducing noise and false positives for accurate predictions. To this end, we first applied various ML strategies, foregoing the use of feature selection, to establish baseline models for CAD prediction utilizing panels of genetic variants. We then compared the predictive performance of these machine learning models to those achieved using PRS-based models. ML-based models were trained on genomic variants as well as on known risk factors to determine the extent to which genomic variants can enhance the predictive power of risk factor-based models. It should also be noted that PRS-based predictions start from the analysis of a pre-selected set of genetic variants. However, in contrast to the ML approach, variants are selected based on association tests, which evaluates each feature individually and selects the ones with the highest statistical significance. In each employed method, the feature selection process commences with a refined set of SNPs, achieved by applying a Minor Allele Frequency (MAF) filter of 0.01 and an R2 threshold value of 0.1 for linkage disequilibrium (LD) to the entire pool of imputed variants (7.87 million). Then, ten fold cross-validation was used to assess model stability and mitigate overfitting by generating 10 training and test data sets. These sets were used to build and evaluate predictive models of CAD based on PRS (PLINK-PRS, LDpred2 and lassosum), and standard ML algorithms coupled with FS approaches. Figure 3 shows the area under the receiver operating characteristic (ROC) curve (AUC) computed on 10 test sets by using CAD-based classification models covering PRS-based strategies (Fig. 3A) and standard ML algorithms working with genotype data (Fig. 3B) or known risk factors for CAD (Fig. 3C). Notably, when excluding risk factors and other relevant covariates, the best methods among PRSs is lassosum, which achieves an AUC of about 0.55. Lassosum uses penalized regression (LASSO) in its approach to PRS calculation. This implies that the best performances for the PRS calculation are achieved by reducing or even eliminating some of the genetic variants selected with association (or univariate) tests. It is also possible to observe that standard ML methods are not able to improve the performance of PRSs. Indeed, the best performing method is LASSO, which also achieves an AUC of about 0.55. However, PRS-based methods exhibit a higher variance on the test sets. This may result from PRS-based predictions being more prone to overfitting compared to the ML approach. Figure 3C shows ML models trained with risk factors achieve high accuracy (AUC > 75%), with LASSO models achieving the best accuracy. PRS and standard ML algorithms do not improve accuracy. SVM with a Gaussian kernel had the lowest performance, as it struggles to model high-dimensional genetic data effectively. In the case of high-dimensional genetic data, the number of dimensions is too high, and the manifold becomes too complex for the kernel to model effectively, resulting in poor prediction performance.
Fig. 3
Classification performance obtained by using PRS, standard classification algorithms and known CAD risk factors. Bar-plots showing the AUC values computed with tenfold cross-validation. Error bars are used to assess model stability, while the different subplots aim to highlight the performance of PRS methods, and ML approaches using genotype data or known risk factors as predictors. A The AUC values obtained by combining PRS scores with logistic regression. B The AUC values obtained by using standard machine learning algorithms. C The AUC values obtained by using known risk factors as input to standard machine learning algorithms
Predicting CAD susceptibility via ML-driven feature selectionAdding genomic variants to traditional risk factors can improve the accuracy of predicting complex diseases such CAD. To this end, different ML-driven feature selection strategies were implemented to assess the effectiveness of various ML-based feature selection techniques in enhancing the accuracy and optimizing the panel size of genomic variants for CAD susceptibility prediction based on genotype SNPs data. Feature selection is important in selecting panels of genetic variants because it helps in reducing the dimensionality of data by selecting a subset of relevant and informative variants. This not only improves the interpretability and understanding of the data, but also enhances the accuracy and performance of predictive models, as it reduces the potential for overfitting and biases. Additionally, feature selection helps in identifying the most important variants that contribute to the phenotype, providing insights into the underlying biological mechanisms. Three different ML algorithms were implemented. The first is based on Random Forest-based feature selection. In this approach, a Random Forest classifier is trained on the data, and the importance of each feature is calculated based on its contribution to the classifier's performance. By considering then top k features based on their importance, it is possible to select a subset of the most informative features for further analysis or to train a predictive model with improved performance. We then implemented a second feature selection strategy using the mRMR algorithm, which can identify a subset of genotype data that is both highly relevant to a phenotype of interest and distinct from other features. It measures both the relevance of each feature with the phenotype and their mutual dependencies and selects a subset of features that maximizes the relevance while minimizing redundancy. Finally, a feature selection strategy based on the result of a standard case/control association analysis using Fisher's exact test implemented in PLINK association analysis was used. The methods generate a score for each variant's contribution to CAD prediction, and different cut-offs were applied to select the top features. These cut-offs were explored to determine the influence of the number of selected features on prediction performance. The feature selection strategy was implemented for each training set of 10-CV and used to train three classification models based on RFs, SVM, and LASSO. Figure 4 shows the performance of ML-driven feature selection algorithms based on AUC values computed with classification models trained with subsets of selected genomic variants (top k) and known risk factors. It is possible to observe that RF-based models systematically outperform LASSO and SVM-based models. Notably, feature selection based on RF classifiers lead to CAD prediction models with an AUC value close to 0.8 (Fig. 4C). This result is achieved by using the top 50 features. Moreover, SVM and LASSO do not perform better than a classification model trained on known risk factors (red dash-dot line) and that SVM-based classifiers achieve high AUC values only when considering large set of features. Finally, we observed a plateau in the classification performance when using the top 50 features selected by the employed feature selection algorithms, possibly indicating that a relatively small set of genetic variants is sufficient for improving risk factor-based models. Figure 5 compares different classification models for CAD prediction, including those based solely on risk factors, all genetic variants (without feature selection), the best performing PRS, which is based on the lassosum, and genomic variants selected through feature selection. Notably, feature selection effectively selects genomic variants enhancing risk factor-based models, with the top 50 variants performing comparably across methods. PRS integration does not increase accuracy, and the best PRS combined with risk factors yields slightly lower performance than top 50 RF-selected features with risk factors.
Fig. 4
Evaluating the accuracy of models that utilize both genotype SNPs and risk factors in predicting CAD susceptibility through various machine learning techniques. Bar-plots showing the AUC values computed with tenfold cross-validation. Error bars are used to assess model stability, while the different subplots aim to highlight the performance of three feature selection strategies. Selected features were systematically evaluated with three different classification algorithms: RF, SVM and LASSO. Each classifier was trained with selected genotypes, known risk factors and PCs. A The AUC values obtained by using GWAS-driven feature selection. B The AUC values obtained by using mRMR-based feature selection. C The AUC values obtained by RF-based feature selection and by selecting the top 50 features. The red dash-dot line represents the classification accuracy achieved by using known CAD risk factors
Fig. 5
Comparing the accuracy of different classification models, feature selection techniques and predictors. Bar-plots showing the AUC values computed with tenfold cross-validation by using two main classification algorithms (RF and LASSO) and different sets of features (or predictors). Feature sets include known risk factors, all SNPs, PRS, and the top 50 genomic variants selected by RF, mRMR and GWAS results. Moreover, classification models were trained with both genotype data and a combination of genotype data and risk factor. Classifiers annotated with top PRS are trained with the best performing PRS method, which is lassosum. Error bars are used to assess model stability
Evaluating the stability of feature selection algorithmsML-driven feature selection strategies were evaluated within a 10-CV framework. We therefore sought to evaluate how frequently the same features or genomic variants are selected across different training sets. Figure 6A displays the consistency of feature selection by depicting the percentage of overlap in the top K features selected across multiple runs of cross-validation. GWAS-based feature selection, which expected to be more robust than RF-based feature selection, since it uses a filtering method for feature selection, it provides the most stable results only when considering the top 10 features. However, when reaching the plateau in the classification performance, which corresponds to the selection of the top 50 features, all feature selection methods exhibit a similar level of model stability.
Fig. 6
Feature selection stability and selection of the most stable genomic variants. A Bar-plots showing the reliability of the feature selection process by visualizing the extent to which the same features were chosen repeatedly across multiple iterations of cross-validation, shown as a percentage of overlap in the top K selected features. B Features that are selected more than 5 times across 10-CV and total number of selected features across the same runs. C Genomic variants that are consistently recognized as significant by different feature selection techniques and are deemed to have a significant impact on the results by all methods used are selected in order to define a small set of novel SNPs for CAD risk prediction
Figure 6B includes detailed information on the number of features that are always selected across the multiple runs of cross-validation. Our subsequent goal was to identify genomic variants that are consistently selected as important by each feature selection strategy and are deemed relevant across all methods. To this end, we identified genomics variants that appear in the top 50 genomic features at least five times, and that are selected across all methods. Figure 6C reports that 6 genomics variants are consistently selected as important by the feature selection strategies.
Assessing the biological relevance of ML-driven variants for predicting CADA literature review was conducted in early 2023 to underscore the significance of the selected genes, and the association between genomic variants and genes was determined using FUMA software version 1.5.2. We identified the top 6 genetic variants that were selected by all feature selection methods and used FUMA [21] to map these genetic variants to their corresponding genes (see Table 2). Selected loci have been described in recent CAD-related GWASs, and the mechanism of action for several of them has been linked either with lipid levels (LDLR, SORT1, and LPA) or with molecular changes within the vascular wall implicated in atherosclerosis (CDKN2A/B, FES/FURIN, PHACTR1) (Additional file 1: Tables S2 and S3). LDLR and SORT1 loci have been associated with changes in low-density lipoprotein (LDL) levels [26, 27], whereas LPA is also associated with lipoprotein (a) levels [28]. On the other hand, the 9p21 locus, harboring ANRIL and CDKN2A/B genes, is one of the strongest genetic associations for CAD identified through GWAS. ANRIL is a long non-coding RNA that regulates gene expression, cell proliferation, senescence, apoptosis, extracellular matrix remodeling, and inflammation [29]. ANRIL exerts its effects through endothelial cell function, macrophage polarization, and VSMC phenotypic transition. ANRIL also regulates plaque stability and is involved in thrombogenesis, vascular remodeling or repair, and plaque stability through its regulation of the tumor suppressor genes, CDKN2A/B. In contrast, the 15q26.1 locus harbors two genes, FES and FURIN, which have been shown to regulate the migration of monocytes and vascular smooth muscle cells and monocyte‐endothelial adhesion, respectively [30, 31]. Finally, the genetic risk locus on chromosome 6p24, which contains PHACTR1 and EDN1 genes, is associated with multiple vascular diseases, including CAD, migraine headache, coronary calcification, hypertension, fibromuscular dysplasia, microvascular angina, and arterial dissection [32, 33]. The expected mechanism of action involves the regulation of vascular smooth muscle cell proliferation and vasoconstriction, as well as the promotion of natriuresis and lower systemic blood pressure through the opposing effects of the ET-A and ET-B receptors. Altogether, this evidence supports that that the ML-selected variants capture widely the mechanistic aspects of the disease etiology, including the risk that arises from lipid levels, inflammation, and vascular biology.
Table 2 Mapping the top 6 selected variants to genes
留言 (0)