
記住我



Chronic rhinosinusitis with nasal polyps (CRSwNP) is an inflammatory disease of the mucosa lining the nasal cavity and paranasal sinuses and is estimated to affect up to 4% of the adult population (1–3). The characteristic nasal polyps (NPs) are benign, edematous, inflammatory lesions that originate from the mucosa of the paranasal sinuses (mainly ethmoid) and can obstruct both the nasal airway and the olfactory cleft (4).
The main symptoms of CRSwNP are nasal congestion, rhinorrhea, and loss of smell (1). Smell loss has a significant negative impact on health-related quality of life (HRQoL) and is reported by patients to be one of the most important and troublesome symptoms of their disease (3, 5–8). Patients with CRSwNP experience a high symptom burden, which significantly impacts both their mental and their physical HRQoL (1, 9). Furthermore, patients often have coexisting inflammatory respiratory diseases, notably asthma and nonsteroidal anti-inflammatory drug-exacerbated respiratory disease (NSAID-ERD) (1). The standard of care for CRSwNP includes intranasal corticosteroids and saline irrigation (3, 10). Sinonasal surgery or oral steroids are needed for patients with uncontrolled and more severe disease. However, recurrence is common, with up to 40% of patients needing further surgery within 18 months (2, 11). Oral steroids may provide short-term relief, although repeated courses are associated with adverse effects including but not limited to weight gain, high blood pressure, and an increased risk of infections.
Although not fully elucidated, it is thought that environmental factors (airborne fungi, superantigenic exotoxins produced by Staphylococcus aureus, and pro-inflammatory biofilms) and individual susceptibility (epithelial barrier dysfunction, innate immunity, and genetics) contribute to the pathogenesis of CRSwNP, with nasal microbiota dysbiosis influencing the inflammatory mechanism determining type 1 or type 2 predominance (12–18). Recent studies have shed further light on the molecular diversity of CRSwNP, with evidence of distinct disease endotypes (19–22). An increased understanding of the molecular mechanisms involved in the pathogenesis of CRSwNP and of the predominant role of type 2 inflammation has led to the development of biologic therapies to block specific type 2 inflammatory pathways in CRSwNP. This review provides an overview of the inflammatory pathways involved in CRSwNP and describes how targeting key drivers of type 2 inflammation is an effective therapeutic option for patients with CRSwNP whose disease is uncontrolled or poorly controlled with standard therapeutic approaches.
2 Immune defense in the airway mucosaThe cells and tissues of the airway mucosal lining act as the first line of defense against airborne pathogens, irritants, and other harmful particles. Penetration of the mucosal barrier that results in infection or damage can trigger an acute inflammatory response involving immune T cell activation and the release of cytokines and other locally acting messengers that induce the local accumulation of immune cells (23–25).
Inflammatory responses can be divided into three types. Type 1 inflammation is primarily involved in host defense against viral, bacterial, and fungal pathogens and is mediated by T helper (Th)1 and Th17 T cells and by type 1 innate lymphoid cells (ILCs). The key cytokines involved in type 1 inflammatory responses include interferon gamma and tissue necrosis factor alpha. Type 2 inflammation is involved in defense against parasitic infections and is also a primary driver of allergic disease. Type 2 inflammation is mediated by various immune cells including Th2 T cells, type 2 ILCs, eosinophils, mast cells, basophils, and immunoglobulin E (IgE)-secreting B cells. The key cytokines involved in type 2 inflammatory responses are interleukin (IL)-4, IL-5, and IL-13. IL-4 and IL-13 are involved in polyclonal IgE formation. IL-4/IL-13 signaling drives class switching of B cells to IgE production (26). Type 3 inflammation is primarily involved in host defense against bacterial and fungal pathogens and is mediated by Th17 cells and type 3 ILCs. The key cytokines involved in type 3 inflammatory responses are IL-17 and IL-22.
3 Pathophysiology of CRSwNPThe chronic inflammatory processes associated with CRSwNP result in cellular changes and structural remodeling of the nasal mucosa (27). These changes consist of morphologic and functional changes to the nasal epithelial cells, including proliferation of basal cells, hyperplasia of mucus-secreting goblet cells, and an epithelial–mesenchymal transition with loss of the differentiation of the ciliated cells at the mucosal surface (27). These changes compromise the barrier function and the regenerative capacity of the nasal epithelium and reduce mucociliary clearance. Degradation of the extracellular matrix also contributes to reduction of the structural integrity of the nasal mucosa, a process that is accompanied by fibrin deposition and tissue edema. Abnormal and excessive fibrin deposition occurs within both the nasal mucosa and the NPs and contributes to the retention of plasma proteins, localized edema, and increased viscosity and adherence of nasal mucus (28). Changes to the neuronal supply also occur, with a decrease in the quantity of olfactory neurons (29) and a switch in olfactory stem cells from neuroregeneration to immune defense (30).
In the West, most patients with CRSwNP display type 2 inflammation, characterized by elevated levels of type 2 cytokines (21). Cluster and gene expression analyses have been conducted to further our understanding of the different endotypes in chronic rhinosinusitis (19–21). One study showed that clusters with a type 2 inflammatory signature were associated with a mixture of phenotypes in which there was 47–64% and 20–37% prevalence of CRSwNP and comorbid asthma, respectively (19). Another cluster analysis identified two clusters that carried a predominantly type 2 signature (which included high levels of IL-5, IL-4, and IL-13), with all patients having CRSwNP, a high frequency of comorbid asthma (>80%), high disease burden including smell impairment, and a high incidence of surgery (20). In a study that used mRNA and protein to identify endotypes in chronic rhinosinusitis, a type 2 signature was predominant in both phenotypes of chronic rhinosinusitis and was associated with loss of smell and asthma comorbidity (21). Type 2 pathophysiology is shared by several other atopic diseases, including asthma, NSAID-ERD, atopic dermatitis (AD), and allergic rhinitis, and these conditions often co-occur. The prevalence of asthma among patients with CRSwNP may be as high as 65%, and the prevalence of NSAID-ERD may be as high as 26% (1, 31). The prevalence of AD among patients with CRSwNP is much lower (9% (31)), although patients with CRSwNP do appear to be at an increased risk of developing AD (32). Almost three-quarters of CRSwNP patients have comorbid allergic rhinitis (31), and up to 86% of patients with CRSwNP are estimated to be sensitized to at least one aeroallergen, with the possibility of symptoms of CRSwNP worsening during allergen season (33).
In East Asia, patients with CRSwNP often display type 1 or a mixed type 1/2 inflammatory pattern, suggesting genetic factors may also play a role (34–36). However, in recent years, there appears to have been a shift toward increasing involvement of type 2 inflammation (19, 22, 37).
3.1 Mechanisms of type 2 inflammation in CRSwNPType 2 inflammation in CRSwNP is characterized by the hyperproduction of IgE, eosinophilic inflammation, and mucus hyperproduction (38). The signaling pathways that initiate these changes are stimulated following injury of the damaged epithelium by pathogens, proteases, and irritants (Figure 1). These insults lead first to the production of the cytokines IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) by epithelial cells, which in turn promote the development of mature Th2 cells. TSLP is the most highly induced of these cytokines and activates type 2 ILCs and mast cells. TSLP also promotes the release of additional type 2 cytokines, notably IL-4, IL-5, and IL-13, thereby amplifying the type 2 inflammatory response. IL-33 mediates eosinophilic infiltration of the nasal mucosa and can also induce IL-4 and IL-5 production from eosinophilic NPs (39, 40). As Th2 cells mature, they too begin to secrete a range of cytokines, including IL-4, IL-5, and IL-13. IL-4 and IL-13 drive the key features of type 2 inflammation, including innate immune cell recruitment to sites of inflammation (41, 42). These immune cells include mast cells, eosinophils, basophils, and M2 macrophages. A key type 2 inflammatory response is B cell differentiation into IgE-producing plasma cells. IgE binding to mast cells induces further cytokine production, especially IL-5, which acts to stimulate eosinophil production in the bone marrow and, together with IL-4 and IL-13, promotes trafficking of eosinophils to sites of tissue inflammation, thereby driving eosinophilic inflammation. IL-4 and IL-13 promote eosinophil trafficking by inducing the production of eosinophil-promoting factors [including IL-5 and eotaxin-1 (43)]. IL-13 also plays a specific role through the stimulation of processes resulting in local tissue remodeling (44–46), including mucus hyperproduction by goblet cells. While under nonpathogenic conditions, these inflammatory responses would be downregulated, patients with CRSwNP continue to have elevated levels of IL-4, IL-13, and IL-5 in both eosinophilic and non-eosinophilic NP tissue, compared with healthy controls (47, 48). Local colonization by S. aureus may play a role in CRSwNP pathogenesis, with enterotoxins secreted by S. aureus acting as superantigens that amplify immune responses and shift inflammation toward the type 2 signature (49, 50). Levels of IgE antibodies to staphylococcal enterotoxin superantigens in NP tissue may be associated with disease severity (51).
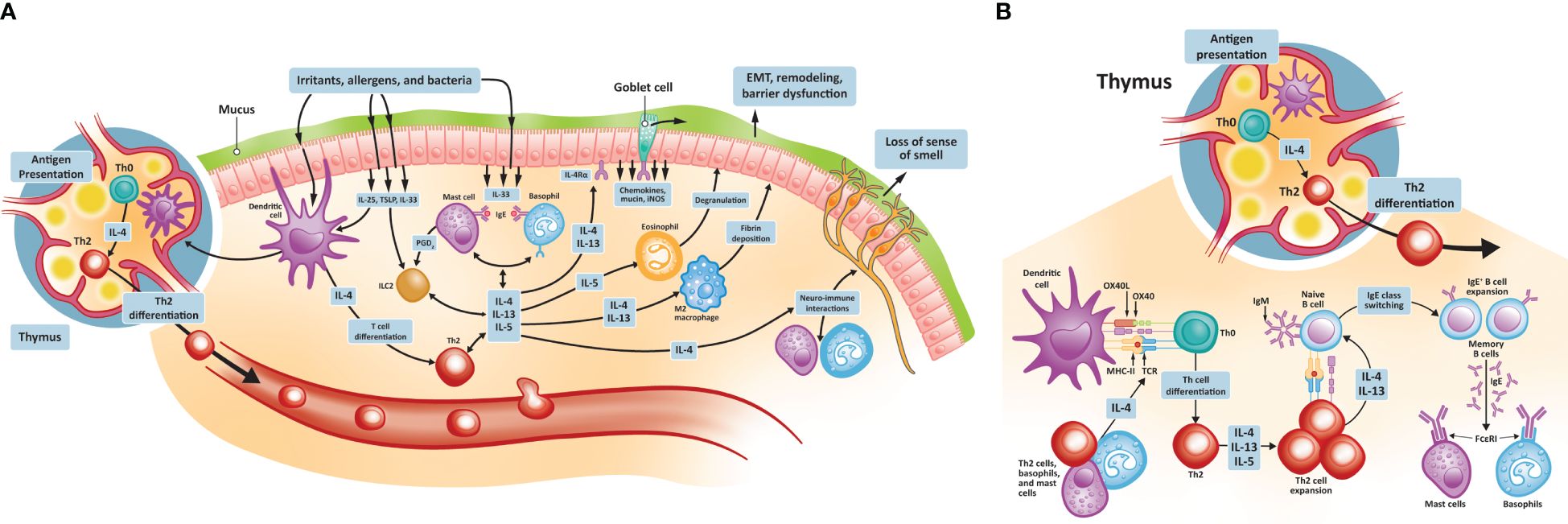
Figure 1 Type 2 inflammatory pathways in CRSwNP. (A) Type 2 inflammatory pathways at the nasal epithelium and thymus; (B) Th2 cell differentiation and activation. CRSwNP, chronic rhinosinusitis with nasal polyps; EMT, epithelial-to-mesenchymal transition; FCεRI, immunoglobulin E receptor; IgE, immunoglobulin E; IL, interleukin; iNOS, inducible nitric oxide synthase; OX40L, OX40 ligand; MHC, major histocompatibility complex; TCR, T cell receptor; Th, T helper; TSLP, thymic stromal lymphopoietin.
Type 2 inflammation contributes to NP formation. IL-4 and IL-13 appear to promote NP formation by increasing the expression of periostin, an extracellular matrix protein involved in fibrotic airway tissue remodeling (52, 53). Periostin is secreted by lung fibroblasts of individuals with asthma and acts as an adhesion molecule, binding other extracellular matrix proteins and resulting in subepithelial fibrosis (53). A similar process of increased extracellular protein adhesion, fibrosis, and consequent edema may be involved in NP formation. Periostin is secreted by nasal epithelial cells and, in addition to promoting tissue remodeling, can stimulate the secretion of TSLP, thereby contributing to the continuation of the exaggerated type 2 inflammatory process (54). Among patients with CRSwNP, the expression of periostin is higher in the NPs of those with coexistent asthma than those without and is positively associated with levels of TSLP (54).
3.2 Chronic inflammation and loss of smell in CRSwNPSmell loss is a symptom associated with considerable decrement to patients’ HRQoL and imposes an increased risk of depression and anxiety (29, 55, 56). Effective treatments are limited, even in the absence of the additional symptoms associated with CRSwNP (56).
Mechanical obstruction of the airflow to the olfactory cleft due to edema, mucus discharge, and/or the physical presence of NPs may contribute to a loss of smell in patients with CRSwNP (29). However, mechanical obstruction is not thought to be the principal cause of loss of smell, and the physical removal of NPs to improve the airflow to the olfactory cleft does not always restore the sense of smell (57, 58). Indeed, patients with chronic rhinosinusitis without NPs may also experience smell loss, indicating the involvement of additional mechanistic factors.
Type 2 inflammation is implicated in the loss of smell through a variety of different mechanisms (29). The histologic changes that result from type 2 inflammation may decrease the number and availability of olfactory neurons and impact their function (59, 60). Recent data support IL-4 playing a direct role in impairing the function of olfactory neurons (61). In mice, IL-4 and IL-13 each increased the calcium uptake in primary olfactory sensory neurons in vitro, while the direct intranasal application of IL-4, but not IL-13, resulted in a rapid loss of smell in vivo (61). These findings suggest nonredundant effects of these cytokines on olfactory function. IL-4-induced loss of smell in this mouse model was attenuated in IL-4 receptor (R)α knockout mice and by treatment with IL-4Rα antibody in wild-type mice. Type 2 inflammation may also impact the formation of new olfactory sensory neurons. Under normal, nonpathogenic conditions, the nasal mucosa is capable of effective regeneration of cells, including olfactory neurons, from horizontal basal cells (30). However, under conditions of chronic type 2 inflammation, the regenerative capacity of the nasal mucosa is lost, as the differentiation of horizontal basal cells is inhibited (30). This direct impact on the formation of new olfactory sensory neurons was demonstrated in a mouse model of Th2‐mediated allergic chronic rhinosinusitis in which a reduction in the number of immature olfactory neurons was observed, while the number of mature olfactory neurons was unchanged (62). Another transgenic mouse model study found that induced expression of IL-13 within the olfactory epithelium resulted in time-dependent loss of neurons from the olfactory epithelium, as well as increased horizontal basal cell proliferation, mucus production, and expression of eotaxin (63). Thus, it appears that chronic type 2 inflammation, typified by elevated IL-4, IL-5, and IL-13, results in impaired olfactory neurogenesis. In addition to type 2 inflammation, there is evidence that type 1 inflammatory cytokines may also contribute to olfactory loss in CRSwNP, indicating a potential role of mixed inflammation (64, 65).
The long-term loss of smell associated with type 2 inflammation in CRSwNP is an area of considerable unmet need, and the potential for recovery of olfactory function is an ongoing area of research (55, 56). As insights emerge about the role of the various signaling pathways of type 2 inflammation, novel targets for therapeutic intervention are becoming apparent, one of which is the IL-4/IL-13 signaling pathway.
3.3 Cytokine signaling pathways in CRSwNPThe earliest components of the cytokine cascade that drives type 2 inflammation in CRSwNP are IL-25, IL-33, and TSLP. These cytokines are released by injured epithelial cells and bind to the respective receptors, namely IL-25R, IL-33R, and TSLPR on the surface of type 2 ILCs (66). Binding to the nascent receptors initiates intracellular signaling, resulting in the production and release of the type 2 cytokines IL-4, IL-5, and IL-13.
The biologic effects of IL-4 are mediated via two types of cell surface receptor. The type I receptor complex binds only IL-4 and consists of an IL-4Rα subunit associated with an accessory common gamma chain. The gamma chain is expressed mainly in hematopoietic cells, with little or no expression on nonhematopoietic cells. The type II receptor complex is a dimeric unit that consists of an IL-4Rα subunit and an IL-13Rα1 subunit and binds both IL-4 and IL-13. IL-13Rα1 is widely expressed in nonhematopoietic cells, although only low levels are expressed by lymphocytes, which predominantly express the gamma chain (67). While IL-13 is able to bind to and initiate intracellular signaling via the type II receptor complex, IL-13 is also able to bind another receptor, IL-13Rα2. IL-13Rα2 is a monomeric receptor that may also act as a scavenger or decoy receptor, preventing the dimerization of the type II receptor complex and acting as a negative regulator of IL-13-mediated signaling (67). The overlapping functions of IL-4 and IL-13 in driving type 2 inflammatory responses are in part due to the shared receptor component, with differences in components of the downstream signaling cascade thought to be responsible for the distinct activities driven by these two cytokines (66, 67). IL-5 binds to IL-5Rα, which forms a complex with a beta chain (66).
Receptor binding of IL-4, IL-13, or IL-5 triggers intracellular signaling cascades mediated by members of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) family of proteins (42, 66). JAK activation results in phosphorylation of specific amino acid residues that then act as docking sites for STATs and other signaling molecules. STATs are soluble transcription factors that, once activated at the receptor, translocate to the nucleus where they induce gene expression. Different STATs drive the expression of different sets of genes; for example, STAT6 increases the expression of the eotaxin-1 gene, which encodes eotaxin, a powerful eosinophil chemotactic protein (68). IL-4 binding to the type I receptor leads to JAK1/JAK3 activation and subsequent activation of STAT6. Binding to the type II receptor by IL-4 (or IL-13) results in activation of JAK1/tyrosine kinase 2 and STAT6 (67, 69). On binding to its receptor, IL-5 activates the JAK1/JAK2 pathway, with subsequent activation of STAT1, STAT5, and STAT3 (66, 70, 71).
4 Therapeutic targeting of the key drivers of type 2 inflammation in CRSwNPSeveral therapies targeting type 2 inflammation are approved or are under investigation for CRSwNP (Table 1). Targeting both IL-4- and IL-13-mediated signaling simultaneously has proved successful and has led to the approval of the first IL-4-/IL-13-targeted biologic agent for the treatment of CRSwNP: dupilumab. Dupilumab is a fully human monoclonal antibody that blocks IL-4Rα, the shared receptor component for IL-4 and IL-13, and therefore prevents signaling by both IL-4 and IL-13 (72, 73). Dupilumab is approved for the treatment of uncontrolled CRSwNP in adults and for other allergic diseases, including certain types of moderate-to-severe asthma, moderate-to-severe AD, eosinophilic esophagitis, and prurigo nodularis (74). Biomarker analysis has revealed that dupilumab treatment in patients with CRSwNP reduced concentrations of type 2 inflammatory biomarkers including eotaxin-3, periostin, eosinophil cationic protein (ECP), and IL-5 in nasal secretions, eotaxin-3, thymus and activation-regulated chemokine, periostin, and total IgE in blood, and leukotriene E4 in urine (75, 76). Dupilumab has also been found to downregulate local IgE production and eosinophil homing (66), and to decrease inflammatory eicosanoid levels and increase levels of anti-inflammatory prostaglandin E2 (77). Dupilumab treatment has been associated with rapid improvement in patient-reported sense of smell (78–80), which suggests the involvement of mechanisms other than improvements in airflow conduction and is consistent with preclinical findings showing direct roles of IL-4 and IL-13 on olfactory function (61, 63).
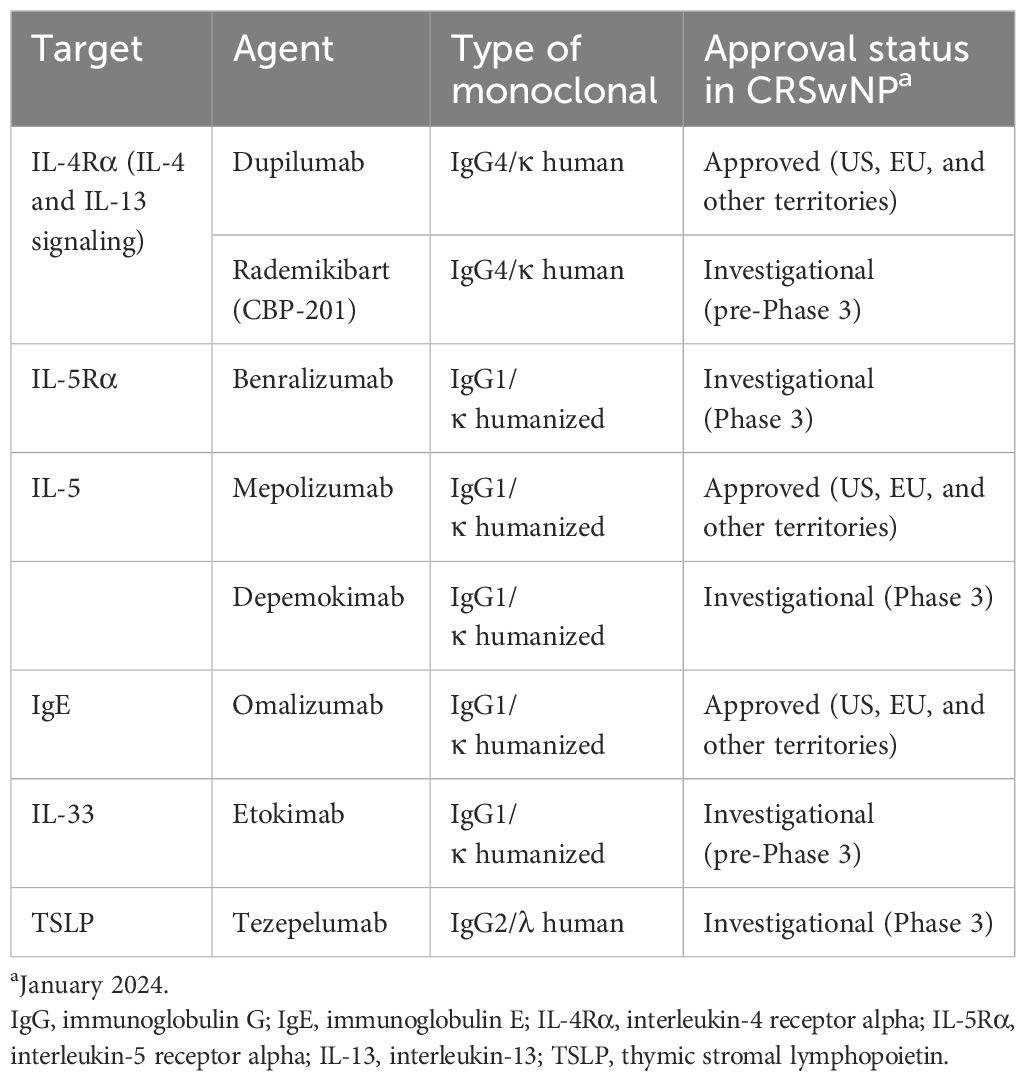
Table 1 Targeted biologic agents for CRSwNP.
Mepolizumab is an IL-5 antagonist monoclonal antibody that was approved first for severe asthma with eosinophilic inflammation. It recently received approval as an add-on maintenance treatment of CRSwNP in adults with inadequate response to intranasal corticosteroids (81). Mepolizumab induced sustained, significant reductions in blood eosinophil counts and reduced ECP and IL-5 receptor alpha levels in serum and IL-5 receptor alpha, IL-6, IL-1 beta, myeloperoxidase, and periostin levels in nasal secretions in patients with CRSwNP (82–84). Benralizumab is a humanized IL-5Rα-directed cytolytic monoclonal antibody that is approved for severe eosinophilic asthma (85) with one Phase 3 study in CRSwNP completed and another ongoing (86, 87). Benralizumab prevents binding of IL-5 to the IL-5Rα subunit and the subsequent heterodimerization with the beta chain subunit required to initiate IL-5-mediated signaling (88). Biomarker findings with benralizumab in CRSwNP are limited but in patients with asthma, benralizumab reduced blood eosinophil counts and reduced serum eosinophil-derived neurotoxin and ECP, and increased serum IL-5, eotaxin-1, and eotaxin-2 (89). Also under phase 3 evaluation is depemokimab, a humanized monoclonal antibody that blocks IL-5 from binding to the IL-5R complex (NCT05274750).
Omalizumab, a monoclonal antibody that targets IgE, is approved by the United States Food and Drug Administration for the treatment of CRSwNP based on the results of the phase 3 POLYP-1 and POLYP-2 studies (90). This agent acts downstream of the cytokine-mediated signaling that drives aberrant type 2 inflammation, acting instead on the effector molecule IgE, preventing the activation of immune cells including basophils and mast cells (91, 92). In patients with CRSwNP and asthma, omalizumab reduced eosinophil counts in blood and nasal smears (93).
Other biologic agents under evaluation for CRSwNP include etokimab (an IL-33-targeted agent) and tezepelumab (an agent that targets TSLP) (91). A phase 2 study of etokimab (NCT03614923) was completed in 2020, although future development plans for this agent have yet to be announced. A phase 3 study is underway for tezepelumab (NCT04851964) and data are expected in 2024. Figure 2 presents a summary of biologics targeting type 2 inflammation in CRSwNP. An investigational IL-4Rα inhibitor, rademikibart (CBP-201), began investigation in a clinical trial in patients with CRSwNP in 2021, but the trial was terminated owing to enrollment challenges (94).
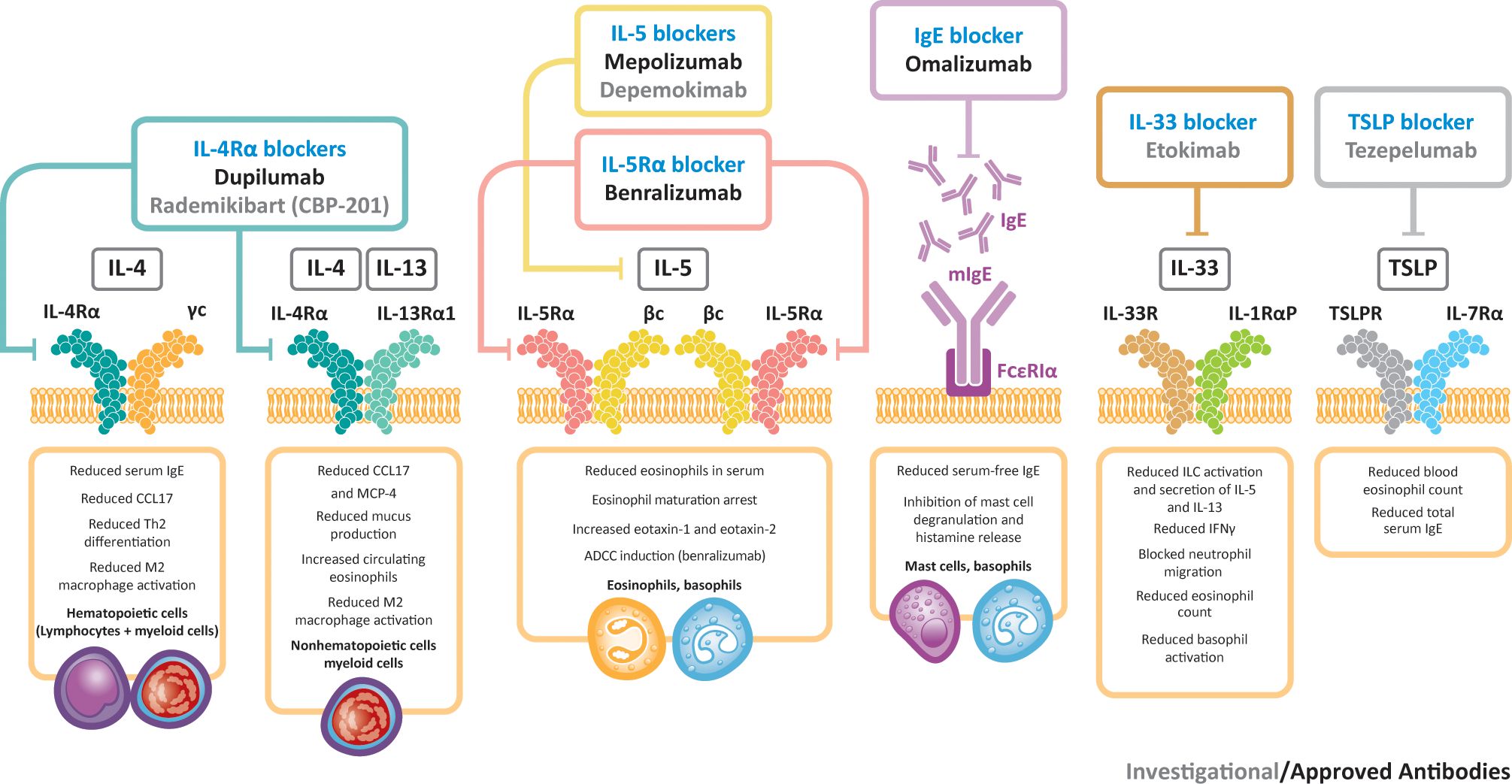
Figure 2 Biologics targeting type 2 inflammation in CRSwNP. βc, beta chain; γc, gamma chain; ADCC, antibody-dependent cellular cytotoxicity; CCL17, C–C motif chemokine ligand 17; IFNγ, interferon gamma; IgE, immunoglobulin E; IL, interleukin; ILC, innate lymphoid cell; MCP-4, monocyte chemoattractant protein-4; R, receptor; Th, T helper; TSLP, thymic stromal lymphopoietin.
To date, no head-to-head clinical studies have been reported in CRSwNP directly comparing the efficacy of biologics targeted at different components of the type 2 inflammatory cascade. However, a recent network meta-analysis of the available evidence on biologics including dupilumab, omalizumab, mepolizumab, and benralizumab found clinically important differences in the effects among these agents: dupilumab ranked among the most beneficial for all outcomes studied (HRQoL, sinusitis symptoms, sense of smell, rescue oral corticosteroids, rescue nasal surgery, nasal polyp score, computed tomography score, and adverse events); with omalizumab among the most beneficial for sinusitis symptoms and HRQoL and intermediately beneficial for sense of smell, rescue surgery, and nasal polyp score; mepolizumab among the most beneficial for sinusitis symptoms and intermediately beneficial for HRQoL, sense of smell, rescue oral corticosteroids, rescue surgery, and nasal polyp score; and benralizumab intermediately beneficial for HRQoL, sense of smell, and rescue oral corticosteroids (95). Limitations of this analysis included small sample sizes and low event rates of rescue sinonasal surgery, rescue oral corticosteroids, and adverse events, which precluded precise effect estimates and high certainty.
5 ConclusionCRSwNP is predominantly a type 2 inflammatory disease. Biologic agents targeting the signaling pathways underlying type 2 inflammation have demonstrated clinical efficacy in patients with moderate-to-severe CRSwNP, reducing symptoms and improving HRQoL.
Author contributionsCB: Writing – original draft, Writing – review & editing. AH: Writing – original draft, Writing – review & editing. SG: Writing – original draft, Writing – review & editing. AP: Writing – original draft, Writing – review & editing. PG: Writing – original draft, Writing – review & editing. SN: Writing – original draft, Writing – review & editing. JH: Writing – original draft, Writing – review & editing. HS: Writing – original draft, Writing – review & editing. JJ-N: Writing – original draft, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study received funding from Sanofi and Regeneron Pharmaceuticals Inc. The funders had the following involvement in the study: funded medical writing support provided by Stephen Whiting, PhD, of Adelphi Group, Macclesfield, UK, in accordance with Good Publication Practice guidance.
AcknowledgmentsThe authors thank Asif H. Khan, MBBS, PhD, MPH, of Sanofi, Bridgewater, NJ, USA for insights and guidance.
Conflict of interestCB is an advisory board member for ALK, ASIT Biotech,AstraZeneca, GlaxoSmithKline, Intrexon Actobiotics, Novartis, Sanofi, and Stallergenes Greer. AH and JJ-N are employees and may hold stock and/or stock options in Sanofi. SG reports advisory board fees from Sanofi and GlaxoSmithKline. AP reports advisory board fees and research support from AstraZeneca, Regeneron Pharmaceuticals, Inc., and Sanofi, and consulting fees and research support from Optinose. PG reports clinical trial funding from and is an advisory board member of 3NT, Argenx, Genentech, Novartis, Regeneron Pharmaceuticals Inc., Roche, Sanofi, and Stallergenes Greer. SN, JH, and HS are employees of and may hold stock and/or stock options in Regeneron Pharmaceuticals Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1356298/full#supplementary-material
Abbreviationsβc, beta chain; γc, gamma chain; AD, atopic dermatitis; ADCC, antibody-dependent cellular cytotoxicity; CCL17, C–C motif chemokine ligand 17; CRSwNP, chronic rhinosinusitis with nasal polyps; EMT, epithelial-to-mesenchymal transition; FCεRI, immunoglobulin E receptor; HRQoL, health-related quality of life; IFNγ, interferon gamma; IgE, immunoglobulin E; IL, interleukin; ILC, innate lymphoid cell; iNOS, inducible nitric oxide synthase; JAK, Janus kinase; MCP-4, monocyte chemoattractant protein-4; MHC, major histocompatibility complex; NP, nasal polyp; NSAID-ERD, nonsteroidal anti-inflammatory drug-exacerbated respiratory disease; R, receptor; OX40L, OX40 ligand; STAT, signal transducer and activator of transcription; TCR, T cell receptor; Th, T helper; TSLP, thymic stromal lymphopoietin.
References2. Chen S, Zhou A, Emmanuel B, Thomas K, Guiang H. Systematic literature review of the epidemiology and clinical burden of chronic rhinosinusitis with nasal polyposis. Curr Med Res Opin. (2020) 36:1897–911. doi: 10.1080/03007995.2020.1815682
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. (2020) 58:1–464. doi: 10.4193/Rhin20.600
CrossRef Full Text | Google Scholar
4. Bachert C, Gevaert P, Holtappels G, Johansson SG, van Cauwenberge P. Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. J Allergy Clin Immunol. (2001) 107:607–14. doi: 10.1067/mai.2001.112374
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Chung JH, Lee YJ, Kang TW, Kim KR, Jang DP, Kim IY, et al. Altered quality of life and psychological health (SCL-90-R) in patients with chronic rhinosinusitis with nasal polyps. Ann Otol Rhinol Laryngol. (2015) 124:663–70. doi: 10.1177/0003489415576181
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Vennik J, Eyles C, Thomas M, Hopkins C, Little P, Blackshaw H, et al. Chronic rhinosinusitis: a qualitative study of patient views and experiences of current management in primary and secondary care. BMJ Open. (2019) 9:e022644. doi: 10.1136/bmjopen-2018-022644
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Mullol J, Mariño-Sánchez F, Valls M, Alobid I, Marin C. The sense of smell in chronic rhinosinusitis. J Allergy Clin Immunol. (2020) 145:773–6. doi: 10.1016/j.jaci.2020.01.024
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Kohli P, Naik AN, Harruff EE, Nguyen SA, Schlosser RJ, Soler ZM. The prevalence of olfactory dysfunction in chronic rhinosinusitis. Laryngoscope. (2017) 127:309–20. doi: 10.1002/lary.26316
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Khan A, Huynh TMT, Vandeplas G, Joish VN, Mannent LP, Tomassen P, et al. The GALEN rhinosinusitis cohort: chronic rhinosinusitis with nasal polyps affects health-related quality of life. Rhinology. (2019) 57:343–51. doi: 10.4193/Rhin19.158
PubMed Abstract | CrossRef Full Text | Google Scholar
11. DeConde AS, Mace JC, Levy JM, Rudmik L, Alt JA, Smith TL. Prevalence of polyp recurrence after endoscopic sinus surgery for chronic rhinosinusitis with nasal polyposis. Laryngoscope. (2017) 127:550–5. doi: 10.1002/lary.26391
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Huntley KS, Raber J, Fine L, Bernstein JA. Influence of the microbiome on chronic rhinosinusitis with and without polyps: an evolving discussion. Front Allergy. (2021) 2:737086. doi: 10.3389/falgy.2021.737086
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Zhu Z, Lan J, Wei R, Xu Y, Hong Y, Bao W, et al. Microbiome and Th cytokines association in chronic rhinosinusitis with or without nasal polyp. Laryngoscope Investig Otolaryngol. (2023) 8:335–45. doi: 10.1002/lio2.1026
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Oakley GM, Curtin K, Orb Q, Schaefer C, Orlandi RR, Alt JA. Familial risk of chronic rhinosinusitis with and without nasal polyposis: genetics or environment. Int Forum Allergy Rhinol. (2015) 5:276–82. doi: 10.1002/alr.21469
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Teufelberger AR, Nordengrün M, Braun H, Maes T, De Grove K, Holtappels G, et al. The IL-33/ST2 axis is crucial in type 2 airway responses induced by Staphylococcus aureus-derived serine protease-like protein D. J Allergy Clin Immunol. (2018) 141:549–59.e7. doi: 10.1016/j.jaci.2017.05.004
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Lan F, Zhang N, Holtappels G, De Ruyck N, Krysko O, Van Crombruggen K, et al. Staphylococcus aureus induces a mucosal type 2 immune response via epithelial cell-derived cytokines. Am J Respir Crit Care Med. (2018) 198:452–63. doi: 10.1164/rccm.201710-2112OC
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Persson EK, Verstraete K, Heyndrickx I, Gevaert E, Aegerter H, Percier JM, et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science. (2019) 364. doi: 10.1126/science.aaw4295
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Tomassen P, Vandeplas G, Van Zele T, Cardell LO, Arebro J, Olze H, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. (2016) 137:1449–56.e4. doi: 10.1016/j.jaci.2015.12.1324
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Turner JH, Chandra RK, Li P, Bonnet K, Schlundt DG. Identification of clinically relevant chronic rhinosinusitis endotypes using cluster analysis of mucus cytokines. J Allergy Clin Immunol. (2018) 141:1895–7.e7. doi: 10.1016/j.jaci.2018.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Stevens WW, Peters AT, Tan BK, Klingler AI, Poposki JA, Hulse KE, et al. Associations between inflammatory endotypes and clinical presentations in chronic rhinosinusitis. J Allergy Clin Immunol Pract. (2019) 7:2812–20.e3. doi: 10.1016/j.jaip.2019.05.009
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Bachert C, Khan A, Lee S, Hopkins C, Peters A, Fokkens W, et al. Prevalence of type 2 inflammatory signatures and efficacy of dupilumab in patients with chronic rhinosinusitis with nasal polyps from two phase 3 clinical trials: SINUS-24 and SINUS-52. Int Forum Allergy Rhinology. (2023) 14:668–78. doi: 10.1002/alr.23249
CrossRef Full Text | Google Scholar
23. Staudacher AG, Peters AT, Kato A, Stevens WW. Use of endotypes, phenotypes, and inflammatory markers to guide treatment decisions in chronic rhinosinusitis. Ann Allergy Asthma Immunol. (2020) 124:318–25. doi: 10.1016/j.anai.2020.01.013
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Vlaminck S, Acke F, Scadding GK, Lambrecht BN, Gevaert P. Pathophysiological and clinical aspects of chronic rhinosinusitis: Current concepts. Front Allergy. (2021) 2:741788. doi: 10.3389/falgy.2021.741788
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Gevaert P, Nouri-Aria KT, Wu H, Harper CE, Takhar P, Fear DJ, et al. Local receptor revision and class switching to IgE in chronic rhinosinusitis with nasal polyps. Allergy. (2013) 68:55–63. doi: 10.1111/all.12054
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Lee K, Tai J, Lee SH, Kim TH. Advances in the knowledge of the underlying airway remodeling mechanisms in chronic rhinosinusitis based on the endotypes: a review. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22020910
CrossRef Full Text | Google Scholar
28. Takabayashi T, Schleimer RP. Formation of nasal polyps: the roles of innate type 2 inflammation and deposition of fibrin. J Allergy Clin Immunol. (2020) 145:740–50. doi: 10.1016/j.jaci.2020.01.027
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Yan X, Whitcroft KL, Hummel T. Olfaction: Sensitive indicator of inflammatory burden in chronic rhinosinusitis. Laryngoscope Investig Otolaryngol. (2020) 5:992–1002. doi: 10.1002/lio2.485
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Chen M, Reed RR, Lane AP. Chronic inflammation directs an olfactory stem cell functional switch from neuroregeneration to immune defense. Cell Stem Cell. (2019) 25:501–13.e5. doi: 10.1016/j.stem.2019.08.011
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Khan AH, Gouia I, Kamat S, Johnson R, Small M, Siddall J. Prevalence and severity distribution of type 2 inflammation-related comorbidities among patients with asthma, chronic rhinosinusitis with nasal polyps, and atopic dermatitis. Lung. (2023) 201:57–63. doi: 10.1007/s00408-023-00603-z
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Hirsch AG, Yan XS, Sundaresan AS, Tan BK, Schleimer RP, Kern RC, et al. Five-year risk of incident disease following a diagnosis of chronic rhinosinusitis. Allergy. (2015) 70:1613–21. doi: 10.1111/all.12759
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Kim DW, Eun KM, Roh EY, Shin S, Kim DK. Chronic rhinosinusitis without nasal polyps in Asian patients shows mixed inflammatory patterns and neutrophil-related disease severity. Mediators Inflammation. (2019) 2019:7138643. doi: 10.1155/2019/7138643
CrossRef Full Text | Google Scholar
36. Wang X, Zhang N, Bo M, Holtappels G, Zheng M, Lou H, et al. Diversity of TH cytokine profiles in patients with chronic rhinosinusitis: a multicenter study in Europe, Asia, and Oceania. J Allergy Clin Immunol. (2016) 138:1344–53. doi: 10.1016/j.jaci.2016.05.041
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Wang W, Gao Y, Zhu Z, Zha Y, Wang X, Qi F, et al. Changes in the clinical and histological characteristics of Chinese chronic rhinosinusitis with nasal polyps over 11 years. Int Forum Allergy Rhinol. (2019) 9:149–57. doi: 10.1002/alr.22234
留言 (0)