Animal experimentation
All animal experiments were approved at RMIT University (AEC#24454) in accordance with the National Health and Medical Research Council of Australia (NHMRC) and ARRIVE guidelines. Male mice were used in this study as longitudinal clinical studies have shown that male sex and childhood asthma were the most significant predictors of abnormal lung function decline later in life [9]. Age matched 3-week-old male C57BL/6 mice were purchased from Animal Resource Centre (Perth, Australia). Mice were first sensitised to house dust mite extract (HDM [D. Pteronyssinus], Stellergenes Greer, US; 100 µg/35 µL) or instilled with saline (SAL) intranasally, which was followed by 4 consecutive daily challenge of HDM (25 µg/35 µL). After the sensitisation period, HDM (25 µg/35µL) was administered once weekly for 8 weeks to maintain chronic allergic airway disease. During this period, mice were also exposed to the smoke of 9 cigarettes/day (CS) or room air as described previously [19].
Tissue collection
Mice were separated into two cohorts for tissue collection (n = 8 per group in both cohorts) and were culled at the end of the protocol via pentobarbital overdose (i.p., 240 mg/kg). For the first cohort of mice, bronchoalveolar lavage (BAL) was performed by flushing the lungs with ice-cold PBS using a 21G canula inserted in the trachea and whole lungs were then collected by carefully removing the trachea and connective tissues. The right superior lobe was prepared for flow cytometry immediately, and the remaining lobes were snap-frozen in liquid nitrogen and subsequently stored at -80 °C. For the second cohort of mice, lungs were inflated with 10% neutral buffered formalin (NBF) at a constant hydrostatic pressure of 25 cm for a minimum of 20 mins. The inflated lungs were excised and further fixed for another 24 h by immersion in NBF with trachea tied.
BAL differential cell count
Total viable cells collected from BAL were calculated using a haemocytometer. Cytospin was then performed, and the slides were stained using the Hemacolor® Rapid Staining Kit (Sigma-Aldrich, US) for differential cell counting [20, 21]. The remaining fluid was centrifuged, and the supernatant (cell-free BAL fluid) was collected and stored at -80 °C for volatile organic compound (VOC) analysis.
Histological assessment of emphysema
Cross sections of the lungs were prepared and stained with haematoxylin and eosin (H&E). Mean linear intercept (Lm) analysis was performed on H&E-stained lung sections that were imaged on an Olympus slide scanner VS120-SS (Olympus, Japan) to determine and quantify emphysema. Five randomly selected fields, at 20× magnification, in the distal regions of each lung section were analysed. One 10 × 10 square grid, with each small square measuring 100 μm × 100 μm, was created and overlaid on an area in each field that avoids the vasculature and airways. The number of alveolar walls intersecting each horizontal grid line was then counted. The Lm was calculated by first subtracting the distance on each horizontal line occupied by any blood vessels and airways from the total length of all horizontal grid lines, then dividing the remaining distance by the total number of alveolar surface intersections counted. The average Lm across all 5 grids was used as the final Lm of each lung sample.
RNA extraction, cDNA conversion and RT-qPCR
Total RNA was extracted from crushed fresh frozen lung tissue using a RNeasy kit according to manufacturer’s instructions (Qiagen, Germany). RNA was then converted to cDNA using a High-Capacity RNA-to-cDNA™ kit (Life Technologies, US). Real time quantitative polymerase chain reaction was then carried out on the Quantstudio™ 7 PCR system (Life Technologies, US) on cDNA samples using the TaqMan™ Fast Advanced Master Mix (Life Technologies, US) with the appropriate primers. Genes were normalised against Gaphd via the delta-delta Ct method as described previously [22, 23].
Flow cytometry
The right superior lung lobes were excised and digested in Liberase TM (Sigma-Aldrich, US) at 37℃ for 45 mins on a shaking incubator. Digested tissue was then passed 5 times through a 21G needle and cells were pelleted by centrifugation at 4 ℃ for 5 mins. Red blood cells were lysed by incubating samples in ACK lysis buffer for 1 min at room temperature, followed by dilution with 10 mL HBSS. Single cell suspension was then obtained by filtering the samples through a pre-wetted 70 μm cell strainer into a 50 mL tube. Spleen samples were isolated by mechanically disrupting the tissue using a syringe plunger on a 70 µM filter and washing with HBSS. Red blood cells were lysed by incubating samples in ACK lysis buffer for 1 min at room temperature, followed by dilution with 10 mL HBSS.
For the myeloid cells, single cell suspensions of lung cells were first blocked with a rat anti-mouse CD16/CD32 antibody (Life Technologies, US) to inhibit non-antigen binding of immunoglobulins to Fc receptors before stained in Fixable Viability Dye (Life Technologies, US) and specific antibodies consisting of PE/Dazzle 594 – CD11b, BV650 – CD11c, AlexaFluor700 – CD45, PE/Cy7 – CD64, AlexFluor488 – Ly6C, BV785 – Ly6G, APC – MerTk, PerCp/Cy5.5 – MHCII (BioLegend, US), PE – Siglec F and BV711 - CD49b (BD Biosciences, US) to analyse leukocyte subsets. Stained cells were fixed using an eBioscience™ IC Fixation kit (Life Technologies, US) and analysed on a BD LSRFortessa™ Flow Cytometer (BD Biosciences, US). A strict gating strategy was used to determine different immune cell populations in single viable cells.
For the lymphoid cells, single cell suspensions of lung and spleen cells were first blocked with a rat anti-mouse CD16/CD32 antibody (Life Technologies, US) before staining in Fixable Viability Dye Near InfraRed (Life Technologies, US) and specific antibodies consisting of PerCP-Cy5.5 –CD3, V450 – CD8, FITC – CD4, PE-Cy7 – CD44 and SB600 – CD62L, to analyse T cell subsets. Stained cells were analysed on a BD LSRFortessa™ Flow Cytometer (BD Biosciences, US).
Volatile organic compound analysis
Snap frozen BALF was thawed on ice. A 450 µL aliquot was transferred into a 20 mL vial and 4 µL of acenaphthene-d10 (concentration 2 µg/ mL) was added as an internal standard. The samples were first agitated at 250 rpm and 80 °C for 10 min and then transferred into a heatex stirrer (set at 1000 rpm and 80 °C) where a solid phase microextraction (SPME) fiber, constituted of Divinylbenzene/ Carbon-Wide Range /Polydimethylsiloxane (DVB/C-WR/PDMS), was introduced into the headspace to adsorb the volatile and semi volatile compounds for 20 min. The SPME fiber was then placed in the gas chromatography’s inlet and allowed to desorb for 1 min.
The gas chromatography mass spectrometer (GC-MS) system used comprised of an AOC6000 autosampler, a 2030 Shimadzu gas chromatograph and a TQ8050NX triple quadrupole mass spectrometer (Shimadzu, Japan). The mass spectrometer was tuned according to the manufacturer’s recommendations using tris-(perfluorobutyl)-amine (CF43). GC-MS was performed on a 30 m GLC Sciences InertCap Pure-WAX column with 0.25 mm internal diameter column and 0.25 μm film thickness. The inlet was set at 250 °C, the mass spectrometer (MS) transfer line at 250 °C and the ion source adjusted to 200 °C. Helium was used as the carrier gas at a flow rate of 1 mL/min. The analysis was performed under the following oven temperature program; 50 °C start temperature, hold for 5 min, followed by a 10 °C/min oven temperature ramp to 250 °C with a following final hold for 10 min. The MS was operated in electron ionisation and MRM (Multiple reaction monitoring) mode. Targeted GC-MS analysis was completed using the Shimadzu Smart Metabolite Database (v1; which covers 496 volatiles, where each target is comprised of a quantifier and qualifier MRM transition. The Resultant data was processed using the Shimadzu LabSolutions Insight software (v4.0), where peak integrations were visually validated and manually corrected where required. 98 annotated metabolites were identified across all groups. All data were analysed in MetaboAnalyst 5.0.
RNA-sequencing
Total RNA was extracted from crushed fresh frozen lung tissue using a RNeasy Plus kit (Qiagen, Germany) according to manufacturer’s instructions, which was then used for bulk RNA sequencing by the Australian Genome Research Facility (AGRF, Melbourne, Australia). Briefly, the purity and integrity of the RNA was first assessed, followed by library construction with a TruSeq Stranded Total RNA kit (Illumina, San Diego, California, US). Twenty million 150-bp paired end reads were performed on the Illumina NovaSeq 6000 platform, and primary sequence data was then generated with the Illumina DRAGEN BCL Convert 07.021.645.4.0.3 pipeline. The raw sequencing data was trimmed to remove low-quality reads using Trim Galore. The cleaned sequence reads were then aligned against the Mus musculus genome (Build version mm39). The STAR aligner (v2.3.5a) was used to map reads to the genomic sequences to generate the raw gene counts.
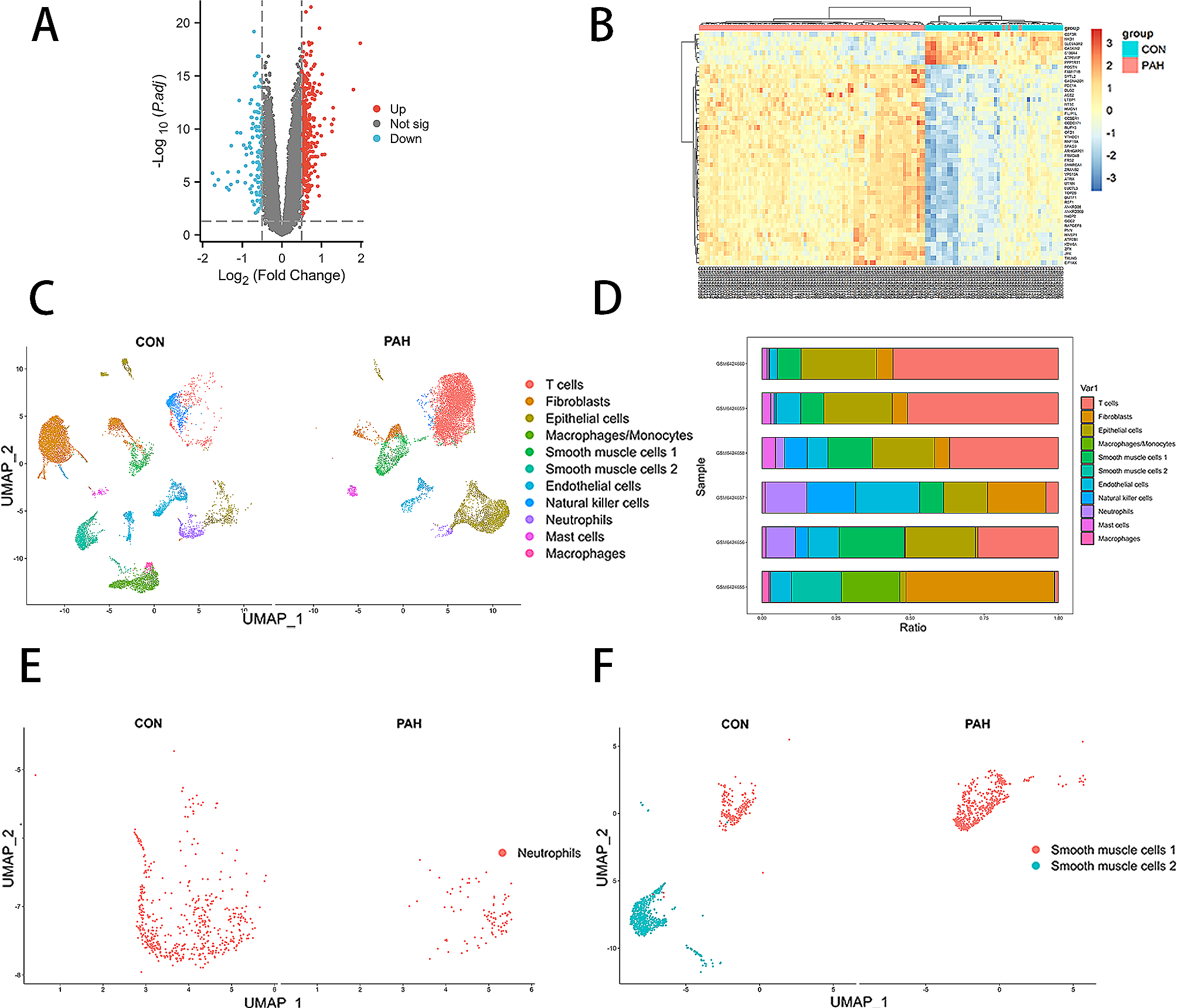
Differentially expressed genes
EdgeR version 3.38.4 was used to identify differentially expressed genes (DEGs) between different groups of comparison. The default trimmed mean of M-values (TMM) normalisation method from EdgeR was used to normalise the counts between samples. A generalised linear model was then used to quantify the differential expression between the groups. DEGs were defined as genes with |logFC| ≥ 1 and false discovery rate (FDR) < 0.05. Visualisation of DEGs on Venn diagram and heatmap was carried out using the R packages ‘ggplot2’, ‘eulerr’ and ‘ComplexHeatmap’. A full list of DEGs is attached in the online file.
Pathway analysis of DEGs
Gene Ontology (GO), Reactome and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DEGs were conducted using R package ‘clusterProfiler’. Reactome pathways and KEGG pathways with p-value < 0.05 and false discovery rate (FDR) < 0.05 were considered significantly enriched. The results were visualized in dot plots using R package ‘ggplot2’.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 9.0 and graphical data are presented as mean ± SEM. Kolmogorov-Smirnov tests were performed to confirm the normal distribution of the data and parametric tests were subsequently used for all analysis. 2-way ANOVA was performed with Tukey’s or Dunnett’s multiple comparisons post-hoc test where appropriate. Statistical significance is declared where p < 0.05 and is indicated with an asterisk (*). (**), (***), (****) are used to indicate p values that are less than 0.01, 0.001, and 0.0001 respectively. All RNAqseq data analyses and visualisation were conducted using R version 4.3.0.





留言 (0)