Chemicals and reagents
Nicotine (GC14043) and 1-methyl-4-phenylpyridinium (MPP+) iodide (GC18188) were purchased from GlpBio (CA, USA). MPTP (CAS:23,007-85-4) was purchased from Aladdin Biochemical Technology Co., Ltd. Ipatasertib (HY-15186; Shanghai, China) was delivered by MedCheExpress. Dulbecco’s modified Eagle’s medium and phosphate-buffered saline (pH 7.4) were obtained from Basal Media (Shanghai, China). The Wisent (Wisent, Canada) company provided Fetal bovine serum. Penicillin–streptomycin solution (100 ×) and trypsin solution were purchased from Biosharp (Hefei, China). Immunostaining fix solution (P0098), immunostaining permeabilization solution containing Triton X-100 (P0096), protease inhibitor cocktail for general use (100× , P1005), phenylmethanesulfonyl fluoride (ST505), phosphatase inhibitor cocktail for general use (50× , P1045), bicinchoninic acid protein assay kit (P0012), radioimmunoprecipitation assay lysis buffer (RIPA, P0013B), sodium dodecyl sulfate polyacrylamide gel electrophoresis loading buffer (P0015), Cy3-labeled goat anti-rabbit IgG (H + L) (A0516), Cy3-labeled goat anti-mouse IgG (H + L) (A0521), fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (H + L) (A0562), and antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (P0131) were purchased from Beyotime (Shanghai, China). The Spark Jade (Qingdao, China) provided SparkZol Reagent (AC0101), SPARKscript II RT Plus Kit (AG0304), and 2XSYBR Green qPCR Mix (AH0104). Antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; P04406) and horseradish peroxidase (HRP)-conjugated secondary antibodies (D110058, D110087) were purchased from Sangon Biotech (Shanghai, China). Anti-tyrosine hydroxylase (TH) (ab137869) and anti-prokineticin 2 (ab76747) antibodies were purchased from Abcam (MA, USA). Cleaved-caspase 3 (Cl-casp3) (Asp175) (5A1E) rabbit mAb (9664) was purchased from Cell Signaling Technology (MA, USA). The PRKAR2A pAb (10,142–2-AP), FoxO3a mAb (66,428–1-Ig), Bax pAb (50,599–2-Ig), and Bcl-2 pAb (26,593–1-AP) were obtained from Proteintech (Wuhan, China). Phospho-Akt-S473 rabbit mAb (AP1208) and pan-Akt rabbit pAb (A18120) were obtained from ABclonal (Wuhan, China). Phospho-FoxO3a (Ser253) rabbit mAb (R24347) was purchased from Zenbio (NC, USA). OMP (sc-365818) was purchased from Santa Cruz Biotechnology (TX, USA).
In vivo experiments
Six-week-old male C57BL/6 mice were provided by the Animal Experiment Center of Anhui Medical University (Hefei, China). These mice were housed with free access to food and water under a 12/12 h light/dark cycle at the room temperature of 24 °C. A randomization number table was used to allocate the mice to different treatment groups. The in vivo experiments were performed according to the regulations suggested in the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Based on the previous experimental results for this platform, the effect size f was 0.85. The two sizes were set at α = 0.05, and the unilateral test efficacy β was 0.9. The number of mice in each group was defined using G*power [21]. The number was calculated to be 7.6 in each group, providing 90% power with a type I error α of 0.05. Therefore, 8 mice were randomly enrolled in each group: normal control group injected with vehicle (Vehicle), small dosage preventative nicotine (0.1 mg/kg daily) group (MPTP + Nic_S), large dosage preventative nicotine (1 mg/kg daily) group (MPTP + Nic_L), large dosage therapeutic (1 mg/kg daily) group (MPTP + Nic_T), and MPTP PD model group (MPTP + Vehicle). MPTP (30 mg/kg) was administered via intraperitoneal (ip) injection for 7 consecutive days [22]. In the preventative treatment group, mice were pretreated with nicotine (ip) 1 week prior to MPTP modeling and then treated with nicotine continuously for 1 week. In addition, therapeutic treatment meant that the mice were administered nicotine (ip) on the first day after the MPTP injection. MPTP was dissolved in saline according to the manufacturer’s protocol. The concentration was set as 6 mg/ mL and the target dosage for each mouse was 30 mg/kg [22]. Nicotine was dissolved in DMSO to form the concentration of 20 mg/100 μL. Then the solution was diluted to 10 mL using saline to 2 mg/mL. This was regarded as the primary nicotine solution and was distributed and stored at − 20 °C. Part of this solution was thawed and diluted to 0.2 mg/mL and 0.02 mg/mL for in vivo experiments. The administration method was intraperitoneal administration as referenced to Liu et al [23]. The volume was determined by the weight of mice before administration. The weight of each mouse were recorded before intervention every day. As to the vehicle administration, similar dilution strategy was employed just without these reagents. And in the morning, nicotine or corresponding vehicle was administrated. In the afternoon, about 6 h later, MPTP was given as described above. Mice were restricted to food and water on the seventh day of the PD modeling. During that time, we only provided basic food (pellets) and water to maintain daily energy consumption. The pellets were intended to be used as incentives in behavioral tests for olfactory function. On the 8th and 9th days, we trained the mice for behavioral tests three times, and the final tests were performed on the 10th day, including the open field, rotarod, and pole-climbing tests for motor function assessment. Pellet-finding experiments and social discrimination tests were also conducted to assess olfactory function in the different groups. Moreover, nicotine positive group was also added to further explore the benefits of nicotine. Thirty-two mice were randomly allocated to Vehicle, Vehicle + Nic_L, MPTP + Nic_L, and MPTP + Vehicle group (n = 8 in each group). And similar intervention strategies mentioned above were employed to treat these mice. Two independent investigators blinded to the group design conducted the behavioral evaluations. All experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Anhui Medical University (No. PJ2023-09-51).
Open field test
The open field test is generally employed to evaluate motor activity and exploration intention in rodents [24]. Mice were placed one by one in the center of the open field arena and allowed to search the environment for a specific period of 300 s. During the test, the animal’s behavior was recorded either manually by an observer or automatically using video tracking software. The total travel distance, central travel distance, and static time were calculated. The trajectory route and heatmap were photographed using an animal behavior analysis system developed by Xinruan Corporation (Shanghai, China).
Rotarod test
The rotarod test provides a measure of motor function and coordination in rodents and is commonly used to evaluate cerebellar function, drug effects, and the impact of neurodegenerative conditions on motor abilities [25]. Mice were trained on the rotarod before the actual test sessions. During training, the rotational speed of the rod was typically constant, and the animals were subjected to multiple trials to improve their motor coordination and balance. Once the animals reached a stable performance level during training, actual test sessions were conducted, often at varying speeds or accelerating rotation (4–40 rpm) for 300 s. The latency was recorded when the mouse fell off the rod. Each mouse was tested and recorded three times. The average time latency for each mouse was calculated.
Pole-climbing test
The pole-climbing test is a behavioral test commonly used in rodent research to evaluate motor coordination, balance, and motor planning [26]. A vertical rough-surfaced rod with a height of 50 cm was prepared. The mouse was placed on top of the rod, typically with its head facing upward. After training for consecutive 2 days, the performance remained stable, and formal experiments were conducted. The latency from the head turning to the front paws touching the ground was recorded. During these behavior tests, it is necessary to avoid physical exhaustion. Therefore, these three exercise experiments were conducted in three days. After each exercise test, these mice will take a rest for 2–4 h for physical recovery.
Buried pellets test
To evaluate the odor detection function, a buried pellet test was conducted, as previously described [23, 27]. Briefly, basic food (pellets) and water were provided during the food and water restriction period. During this time, the mice were allowed to familiarize themselves with the pellet. In this experiment, mice tended to have an incentive to find buried pellets. The pellet was randomly buried 0.5–1 cm below the padding at the corner of the test cage (45 × 24 × 20 cm). The mouse was initially placed at the center of the cage. The latency to find the pellet and start eating was recorded. The total duration of the experiment was 5 min. If the mouse did not find the pellet within 5 min, the time was recorded as 5 min. Between different trials, the padding was changed to avoid scent disturbances between the mice.
Social scent discrimination experiment
The social scent discrimination trial was designed based on previous studies [23, 28]. Briefly, cubic wood (2 × 2 × 2 cm) was incubated with 20 g of mouse padding from the tested mice for 24 h. Mice were habituated to their cages for 1 h without water or food. The mouse was then tested in a place with two wooden cubes (one with its own smell and the other with an unfamiliar smell). Sniffing behaviors were recorded, and the ratio of time spent on each wood cube was calculated and analyzed.
After behavioral evaluation, half of olfactory tissues of three mice in each group were sent for RNA sequencing. The other half of these olfactory tissues were used for RT-qPCR. The half of olfactory tissue of another three mice were employed for Western blot. The other half of these three mice were applied for IHC and IF staining. The remaining two brain tissues were stored in − 80 °C for additional use.
Transcriptional RNA sequencing
The tissues of mice OBs were collected, immersed in RNAlater RNA Stabilization Reagent (Qiagen, 76,104), and stored at − 80 °C. Transcriptional RNA sequencing was performed by the LC-bio Corporation (Hangzhou, China).
SYBR Green reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the OB using the SparkZol Reagent (AC0101), according to the instructions of manufacturer. The corresponding cDNA library was obtained using a SPARKscript II RT Plus Kit (AG0304) for reverse transcription. Real-time qPCR was conducted employing 2XSYBR Green qPCR Mix (AH0104). GAPDH was used as a reference to normalize prok2R expression. The quantitative analysis was performed using the 2−△△Ct method. The primers were provided by Sangon Biotech. These primers include the following sequences: prok2R: forward, 5′-TACCAACCTCCTCATTGCTAACC-3′; reverse, 5′-GTGGTTTCAAAGGGTGGACAATAG-3′; GAPDH: forward, 5′-CAGTGGCAAAGTGGAGATTGTTG-3′; reverse, 5′-TCGCTCCTGGAAGATGGTGAT-3′.
Western blot (WB) assay
Treated cells and brain tissues were harvested and stored at − 80 °C. Then RIPA lysis buffer supplemented with fresh protease inhibitor cocktail, phenylmethanesulfonyl fluoride, and phosphatase inhibitor cocktail was used to lyse the cells or brain tissues. The lysates were sonicated using an ultrasonic cell breaker. After incubation on ice for 30 min, the samples were centrifuged at 12,000 rpm for 30 min. The supernatants were collected and quantified using a bicinchoninic acid assay kit (P0012). A loading buffer was added, and the mixture was boiled in a metal bath for further use. Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to separate the proteins. The extracted protein samples were added into each well, condensed in a 4% stacking gel, and separated in an 8–12% resolving gel under appropriate electric fields. After electrophoresis, the proteins were transferred (blotted) from the gel onto a polyvinylidene fluoride membrane. To reduce the nonspecific binding of antibodies and decrease background noise, the membrane was blocked with a blocking solution containing bovine serum albumin (BSA). A specific primary antibody that recognized the target protein of interest was added to incubate with the membrane overnight at 4 °C with gentle shaking. During this process, the primary antibody binds to the target protein to form an antigen–antibody complex. The TBS-T was used to wash the membrane three times to remove any unbound or nonspecifically bound antibodies. The corresponding secondary antibody was then added and incubated with the membrane to form a sandwich-like structure around the target protein. Finally, the Tanon 5200 system (Tanon Technology Co., Ltd.) was used to capture protein band signals from the membrane.
Immunohistochemical (IHC) and Immunofluorescence (IF) staining
IHC staining was performed as previously described [29]. 10% neutral-buffered formalin was employed to fix the samples for 24 h. Samples were embedded in paraffin, and standard coronal brain sections (40 μm) were sliced. Paraffin-embedded tissue sections were deparaffinized by heating and treated with xylene to remove wax. The sections were rehydrated using a series of graded alcohol washes (anhydrous, 85%, and 75% ethanol). The citrate buffer (10 mM sodium citrate, pH 8.5) at 90 °C for 30 min was employed to retrieve antigen. PBS was used to wash the sections for three times. The blocking buffer containing 3% BSA, 0.1% Triton X-100, and 0.05% Tween 20 in PBS was used to permeablize these sections. Primary antibodies against TH (rabbit pAb, 1:500) and Cl-casp3 (rabbit mAb, 1:500) were incubated with the slices overnight at 4 °C. After three times of washes with PBS, the Alexa dye-conjugated secondary antibodies (for IHC) or FITC/Cy3-labeled secondary antibodies (for IF) were added to incubate with these sections for 1 h around 25 °C. Hoechst stain (for IHC) or antifade mounting medium with DAPI (for IF) was added to the sections and incubated for 5–10 min at 25 °C to label the nuclei. The slices were evaluated under the XYZ motorized stage connected with Olympus OX41 microscope and counted by a researcher who was blinded to the grouping information using the Optical Fractionator probe of Stereo Investigator software [30, 31]. The quantified striatal TH-positive terminal density was analyzed by densitometry using Image-J software.
In vitro experiments
Human embryonic kidney 293 T (HEK293T) cell line and primary mouse olfactory neurons (CP-M145) were purchased from Procell (Wuhan, China). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution in an incubator (pO2, 21%; 5% CO2). Primary mouse olfactory neurons were incubated in the specific medium provided by the manufacturer.
Cell viability experiment
A Cell Counting Kit-8 (CCK8) assay was used to assess the cell viability. A 96-well plate was seeded with 5000 cells per well and cultured for 24 h to a cell confluence of approximately 60–70% per well. Subsequently, gradient concentrations of nicotine and MPP+ were added. After incubation for 24 h, CCK8 solution (10 μL) was added into each well and incubated for 2 h. Finally, a microplate reader was used to record the at 450 nm.
Live/dead staining
HEK293T cells were seeded in 6-well plated and incubated for 24 h. We then employed different treatment strategies and classified them into several groups: group 1, normal control; group 2, nicotine treatment group; group 3, nicotine + MPP+ treatment group (nicotine was added 4 h before MPP+ treatment); and group 4, MPP+ treatment group. After incubation for 24 h, the cells were washed and incubated with calcein AM and propidium iodide for 30 min. The cells were then gently rinsed three times with PBS to remove the dye. Afterward, these treatment groups were photographed using an inverted fluorescence microscope (ECLIPSE Ti2; Nikon, Japan).
Analysis of apoptosis using flow cytometry
The cells in the different groups were collected, washed, and resuspended in the medium provided in the Annexin-V and PI Apoptotic Assay Kit. The cells were then stained with annexin-V and propidium iodide solutions. CytoFlex (Beckman Coulter) was used to calculate and analyze the number of apoptotic cells in different groups.
Transmission electron microscope (TEM) analysis
Cells from the different groups were collected and fixed using 2.5% glutaraldehyde. The fixed samples were dehydrated using a series of alcohol or acetone washes to remove water and prevent electron-beam scattering during imaging. The dehydrated samples were infiltrated with an epoxy resin to provide structural support and facilitate thin sectioning. The resin was then polymerized to form a solid block. Ultrathin sections (70–90 nm thick) were cut from the embedded blocks using an ultramicrotome. Sections were mounted on a TEM copper grid. After staining with uranyl acetate and lead citrate, TEM imaging was performed using Hitachi-7800.
Construction and transfection of lentivirus
Lentiviruses containing prok2R shRNA and non-silencing control shRNA (shNS) were constructed and provided by GeneChem (Shanghai, China). Similarly, prok2R overexpression (prok2R+) and negative control (prok2Rvector) lentiviruses were also produced. The shRNA target sequences were designed as follows: shNS, 5′-TTCTCCGAACGTCACGT-3′; prok2R-RNAi (shProk2R#1), 5′-GCTGAGACCTATAACCCTGAT-3′; prok2R-RNAi (shProk2R#2), 5′-GCAGATTTAATAGACGAGTAT-3′; prok2R-RNAi (shProk2R#3), 5′-GAAAGGATAGTCAAAGCTGAT-3′. HEK293T cells were transfected according to the manufacturer’s instructions. Then the infected cells were incubated with puromycin (2.0 µg/mL) to screen the successfully transfected cell lines. Finally, the WB was used to evaluate the silencing efficiency of the target genes.
Confocal laser scanning microscopy (CLSM) analysis of cells
HEK293T cells or primary olfactory neuron-climbing sheets were cultured for 24 h. The sheets were then washed, fixed, and permeabilized. We used 3% BSA to block the antigen for 1 h. Then, the primary prok2R, OMP, and Cl-casp3 antibodies were incubated with the sheets overnight at 4 °C. The sheets were washed with PBS and incubated with FITC/Cy3-labeled secondary antibodies for 1 h. The sheets were then observed, and images were captured using Zeiss LSM880.
Statistical analysis
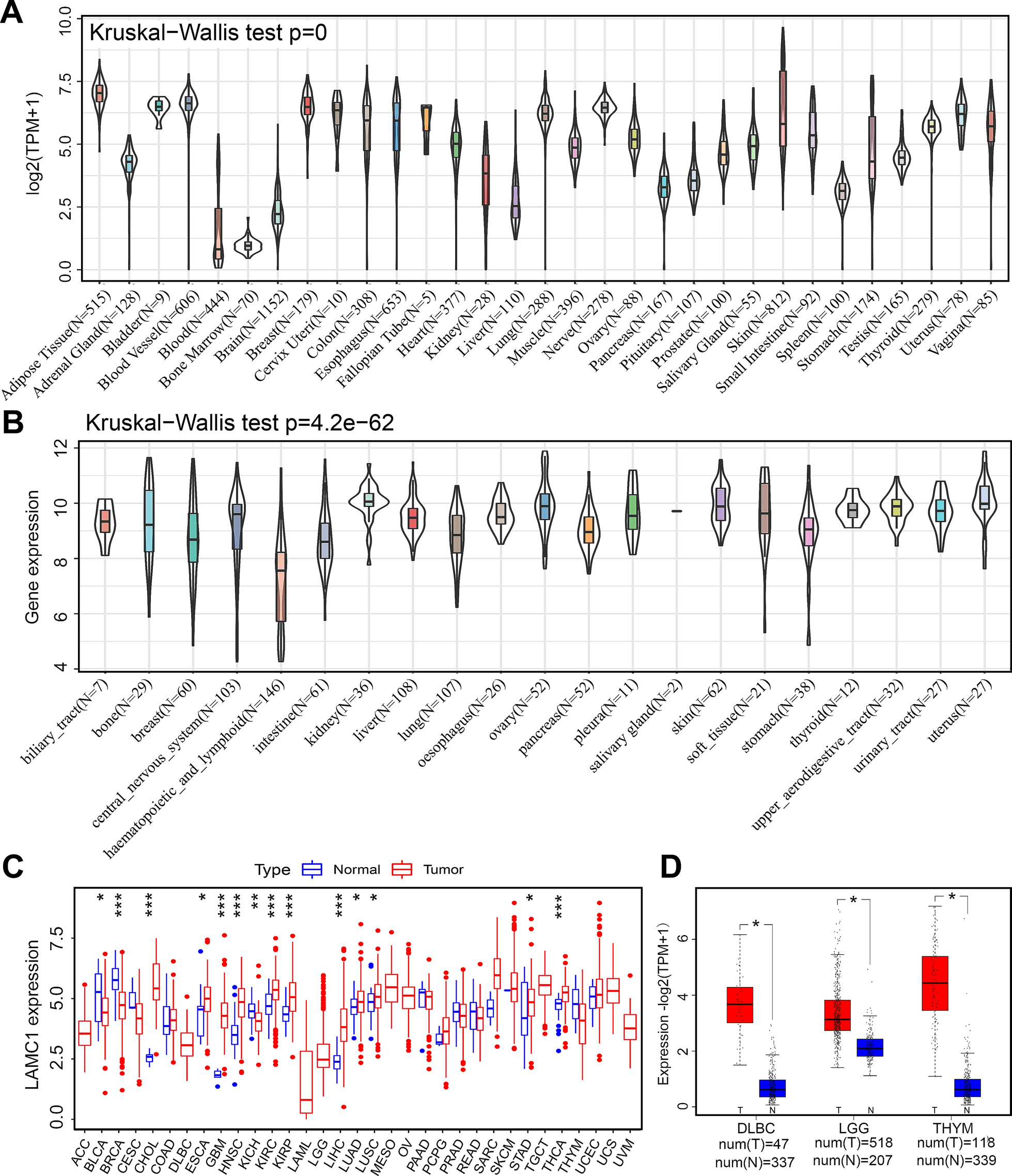
The SPSS software (version 19.0; IBM, Armonk, NY, USA) and GraphPad Prism (8.0.2) software were employed to calculate statistical analyses. Data were presented as the mean ± standard deviation, unless indicated otherwise. Data distribution normality was determined by Shapiro–Wilk test. If the data distribution was normalized distributed, Student’s t-test and one-way analysis of variancewere used to evaluate continuous data comparing the differences between two groups and across multiple groups, respectively. Otherwise, Kruskal–Wallis test could be adopted to conduct further analysis. Bonferroni corrections were employed to control type I error for normalized distributed data. False discovery rate (FDR) comparison corrections were applied to reduce type I error for non-normalized distributed data. The two-sided adjusted p-value less than 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001).





留言 (0)