
記住我



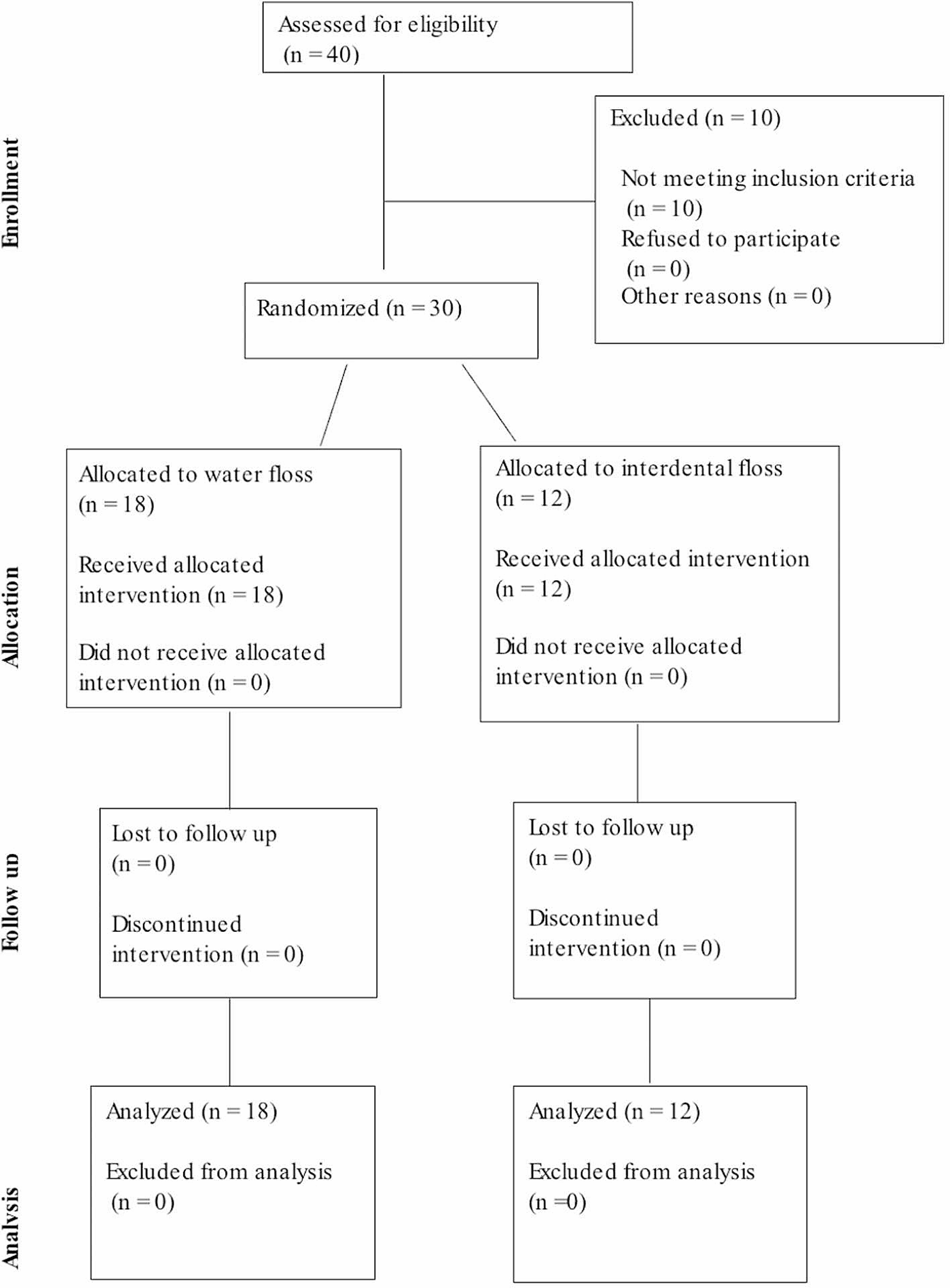


The search resulted in 116 publications from Embase, Web of Science Core Collection, and Scopus. Following title and abstract screening, 11 publications were included for full-text screening. Two publications referred to the same study so were therefore considered as one. Ten studies were included for qualitative analysis (Fig. 1).
Fig. 1
Flow chart indicating the number of records identified and included for qualitative analysis on developmental defects of enamel in people with cystic fibrosis
Study characteristicsOf the ten studies included, three were conducted in the USA [6,7,8], five in Europe [9,10,11,12,13], one in Brazil [14],and one in Turkey [15]. A total of 476 people with CF were studied. The earliest study included was published in 19766, with the most recent study published in 201912.
Eight studies included children and adolescents with CF up to the age of eighteen6,8–11,13−15. One study included both children and adults with CF over the age of eighteen [7]. One study limited study participants to adults with CF over the age of eighteen [12].
Several different clinical indices were used to categorize enamel defects. Four studies used the modified developmental defects of enamel (mDDE) index [8, 10,11,12], and one study used the DDE index [14]. The study conducted by Narang [9] categorized enamel defects based on WHO criteria. Primosch [7] measured defects using criteria developed by Russel [16], whereas Peker used criteria developed by Weerheijm [17]. The study conducted by Jagels and Sweeney [6] measured enamel defects as a percentage of teeth showing hypomineralisation.
Eight studies included control groups [6, 7, 9,10,11,12, 14, 15], and only two studies did not [8, 13]. Six studies included age and gender-matched ‘healthy’ individuals in the control group [7, 10,11,12, 14, 15], one study included non-CF siblings in the control group [6], and another study included control subjects with chronic respiratory conditions [9]. This data is summarised in Table 1.
Table 1 Tabulated Results showing the Author, Year of Study, Number of Individuals Examined, Age Group, and Enamel Defects MeasurementFour studies reported a statistically significant increase in the prevalence of DDE in PwCF compared to non-CF control groups [8, 10,11,12]. The study by Narang [9] reported a statistically significant difference in the prevalence of DDE in the CF control group aged 6–9 years only. Two studies reported a non-significant increase in the prevalence of DDE between CF a non-CF control groups [14, 15]. Primosch [7] found that DDE were more common in people with CF compared to healthy individuals. Two studies by Jagels and Sweeney [6] and Collard [13] reported no difference in the prevalence of enamel defects between PwCF and non-CF people/ national averages. These results are summarised in Table 2.
Table 2 Data abstracted from studies regarding developmental defects of enamel in People with Cystic FibrosisRisk of BiasThe risk of bias was assessed using several headings. These were adapted from previous reviews in this subject field by Chi et al. [18] and Coffey et al. [19]. This assessment was conducted by two investigators working independently (FO’L & NC). Any conflict was reviewed by a third investigator (MH). Studies meeting all seven criteria were classified as being at low risk of bias. Studies meeting three to five criteria were classified as medium risk of bias. Studies meeting less than three criteria were classified as being at high risk of bias. Seven studies were classified as being at medium risk of bias, and three as a high risk [6, 7, 13]. Eight studies included a control group [6, 7, 9,10,11,12, 14, 15]. Only one study provided an explanation for the study group [6]. The study by Jagels and Sweeney included non-CF siblings [6] in the control group. The study by Narang included individuals with chronic respiratory illness as control subjects [9]. The inclusion of such control groups may influence results as siblings may be carriers of the CFTR mutation, and people with respiratory illness may have a similar therapeutic history to PwCF. A limitation common to all studies was the absence of examiner blinding. The authors appreciate this may be difficult to achieve owing to the complex medical history of PwCF. Five studies were conducted by one examiner and did not provide sufficient information regarding examiner reliability [6, 7, 9, 14, 15]. Seven studies adopted standard defect measures (e.g., DDE, mDDE) [8,9,10,11, 13, 14], while three studies did not (Table 3) [6, 7, 15].
Table 3 Risk of bias assessment
留言 (0)