
記住我








Coilia nasus, namely taper-tail anchovy, is a small-to medium-sized fish that can be divided into two stocks based on their life stages. One stock resides in the ocean and migrates to freshwater for reproduction, which is widely distributed in the coasts of the Northwest Pacific, including Korea, China, and Japan. In China, the migratory stock is mainly fished from the Yellow Sea and East China Sea (Ma et al., 2023). In spawning season, Coilia nasus adults aggregate in the estuary of the Yangtze River and then migrate to the middle and lower reaches of the Yangtze River for reproduction before their final gonadal maturation. The other is a freshwater-resident stock which spends its whole life cycle in the lakes in the lower reaches of the Yangtze River, and becomes the most dominant species in the lake ecosystem. It might be possible that chromosomal inversions caused the differences in morphology, physiology, and behaviour between the anadromous and resident stocks in the face of environmental heterogeneity in C. nasus (Zong et al., 2021). Over the past few decades, serious pollution, overfishing, and changes in aquatic ecology have resulted in a dramatic decline in populations of the migratory stock, which has a high economic importance in China (Zhang et al., 2005). However, C. nasus is highly responsive to stress, and this often causes death as soon as it is out of the water, which obstructed artificial breeding via capturing wild sexually mature individuals (Du et al., 2014). The Yangtze River to artificial pond diversion project was applied to capture wild juveniles of C. nasus (Zhang et al., 2006), therefore the tapertail anchovy was reared and domesticated in an artificial pond, and bred for five to six offspring to date.
The artificially reared taper-tail anchovy shows an evident phenomenon of population-asynchronous spawning, and the reproduction of tapertail anchovy continues to occur from April to July. Some three-year-old individuals have an asynchronous ovary development, although they breed in stable aquaculture conditions including nutrients, water temperature and environmental factors (Du et al., 2017). A best way to understand the mechanism of fish reproduction is to explore the gene regulation of ovarian development (Hayes, 1998). In teleost fish, reproduction is closely regulated by the hypothalamus-pituitary-gonadal (HPG) axis (Sofikitis et al., 2008). Gonadotrophin-releasing hormone (GnRH) in the hypothalamus, gonadotropin hormones (GHs) in the pituitary, and sex steroid hormones in the gonads have a pivotal role in the endocrine regulation of fish reproduction (Liu et al., 2013), resulting in complex reproduction modes, such as gonochorism (Jalabert, 2005), unisexuality (Schartl et al., 1995), and hermaphroditism (Avise and Mank, 2009). In general, fish sex determination is forced by genetic and/or environmental factors (Muralidhar and Veller, 2018; Bókony et al., 2019; Strüssmann et al., 2021). Genes in regulating sex determination and development exhibit spatio-temporal specificity in various species (Sinclair et al., 1990). Amh (anti-Müllerian hormone), Wnt (wingless-related integration site), and dmrt (Doublesex and mab-3-related transcription factor) have been identified to be involved in sex determination and sex differentiation processes (Dong et al., 2020; Farhadi et al., 2021; Liu et al., 2022). In addition, the biological signaling pathways are involved in steroidogenesis to mediate the gonad development of teleost fish, such as estrogen and TGF-β signaling pathway (Baroiller and Guiguen, 2001; Amberg et al., 2013). Recently data from taper-tail anchovy showed that several genes involved in the sex differentiation cascade exhibited characteristic expression in both gonadal somatic cell lines cultured in vitro (Kan et al., 2022) and in spermatogonial stem cell lines under long-term in vitro culture conditions (Gu et al., 2023).
RNA-seq technology has been widely used to profile genes involved in gonad development and their molecular mechanisms in fish (Du et al., 2017). Gender-specific transcriptomes have revealed a number of genes involved in gonad development, including medaka (Oryzias latipes) (Dechaud et al., 2021), threespine (Gasterosteus aculeatus) (Kaitetzidou et al., 2022), Amur sturgeon (Acipenser schrenckii) (Zhang et al., 2016), red-tail catfish (Hemibagrus wyckioides) (Wei et al., 2023) and Coreoperca whiteheadi (Liu et al., 2023). Transcript profiles were generated from sexually mature and immature gonads of fish species to identify sex-associated molecular markers (Amberg et al., 2013) and sex-related genes (Cao et al., 2022). High quality transcript assemblies were obtained from known reference genomes of the species. The whole genome sequences of taper-tail anchovy have been published (NCBI PRJNA422339) (Xu et al., 2020), and a gap-free reference genome sequence of anadromous C. nasus was available (SRP405363) (Ma et al., 2023). In the present study, gene expression profiles were first generated from the brain and pituitary tissues of female C. nasus at ovarian stages II and IV, respectively, under the same aquaculture conditions. Differentially expressed genes (DEGs) between ovary-stage II brain/pituitary and ovary-stage IV brain/pituitary were detected. Important candidate genes and pathways involved in ovary development were then identified. Weighted gene co-expression analysis (WGCNA) was comprehensively performed to discover the transcriptional network and the regulatory hub genes in response to ovary development. This study aims to detect gene coordination patterns in the hypothalamus-pituitary-gonad axis in C. nasus with unsynchronized ovary development and provides a theoretical basis for future artificial breeding.
2 Materials and methods2.1 Experimental fish collectionSix healthy female C. nasus were collected simultaneously from Jiangzhiyuan Farm, Jiangsu Province, China, during the breeding season. Gonadal development was observed by anatomy and paraffin section, and three individuals with ovarian stage Ⅱ and three with stage Ⅳ were selected. Brain and pituitary samples were dissected and immediately stored in liquid nitrogen until the total RNA was extracted.
2.2 RNA extraction, library construction and sequencingTotal RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according the manufacturer’s instructions. RNA concentration was determined using a NanoDrop-2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) and RNA integrity was determined by agarose gel electrophoresis. Poly(A) mRNA was isolated from the total RNA using Oligo (dT) and cleaved into short fragments using a Covaris S220 Focused-Ultra sonicator (Covaris, California, USA) and then used as templates for the synthesis of first- and second-strand cDNA according to the protocol of the Super Script Double-Stranded cDNA Synthesis Kit (Thermo Fisher Scientific, MA, USA). The cDNA was purified using a QIAquick PCR Purification Kit (QIAgen, Düsseldorf, Germany). After end repair, poly(A) addition and sequencing adapter ligation, the optional 300–400 bp fragments were selected by agarose gel electrophoresis, and enriched by PCR amplification to construct the cDNA library, followed by sequencing and generation of 150 bp paired-end reads using the Illumina HiSeq Ten platform. Three independent biological replicates were performed for stage Ⅱ and Ⅳ based on each tissue.
2.3 Read processing, reference sequence alignment, alternative splicing and novel transcript predictionRaw reads were filtered using fastp v0.23.4 (https://github.com/OpenGene/fastp) to obtain high quality clean reads, and reads containing adaptor sequences, N% greater than 10%, and Q20% less than 20 were excluded. Clean reads were aligned to reference sequences (SRP405363) (Ma et al., 2023) using hisat2 (Shibaguchi and Yasutaka, 2022). The mapped results were used to determine alternative splicing occurring at different developmental stages using rMATs v4.0.2 (http://rnaseq-mats.sourceforge.net/rmats4.0.2/index. html). Transcripts for each sample were assembled using StringTie v2.2.2 (Pertea et al., 2015) and then merged into a set of transcripts for all samples to predict novel transcripts by comparison with annotation using gffcompare v0.11.2 (Pertea and Pertea, 2020).
2.4 Gene expression and differential expressed gene functional annotationGene expression counts were obtained from each sample via clean reads aligned to the reference using bowtie2 v2.5.2 (Mortazavi et al., 2008). Fragments per kilobase of transcript per million mapped reads (FPKM) values for the gene expression levels were calculated using eXpress v4.18.2 (Pertea et al., 2015). Relativity of biological replicates was calculated using the Pearson correlation coefficient between samples. Based on the gene expression level, differentially expressed genes were identified using DESeq2 v3 (Love et al., 2014). Thresholds for significant differential expression were set as a p-value <0.05, FDR (false discovery rate) ≤ 0.05, and absolute of log2 (fold change) ≥ 2. Finally, the differentially expressed genes were used for GO and KEGG enrichment analysis.
2.5 Identification of co-expressed network genes and hub genesWGCNA analysis was used to assess the co-expressed gene clusters of brain and pituitary during the ovarian developmental periods using the OmicShare tools, a free online data analysis platform (https://www.omics.hare.com/tools). These expressed genes were clustered according to their log2 (FPKM) values. Pearson’s correlation coefficient was calculated for all genes obtained from brain and pituitary with ovarian developmental stage Ⅱ and stage Ⅳ, and a soft-threshold power (β = 8) was chosen to obtain a topological network. The hub genes in each module were identified by the value of kME (Module Eight Gene-based Connectivity).
2.6 qRT-PCR validation of differentially expressed genesThe qRT-PCR method was used to verify the differential expression of genes. Seventeen differentially expressed genes were selected from the brain and pituitary, and the β-actin gene was used as a control. The qRT-PCR primers were designed using the Primer Premier 6 (Bustin and Huggett, 2017), listed in Table 1, and synthesized by Sangon Biotech (Shanghai, China). The qRT-PCR was performed on an ABI 7500 Real-Time PCR System (ABI, New York, USA) using the 2−ΔΔC method (Livak and Schmittgen, 2001). The reaction mixture consisted of 1 μL cDNA (60 ng/μL), 10 μL qPCR SYBR Green Master Mix (Hieff, Bioscience Inc., Hamburg, Germany), 0.5 μL of each primer, and 8 μL milli-Q water. Reactions were performed at 95°C for 30 s; 35 cycles of 95 °C for 5 s, 60°C for 20 s and 72°C for 10 s. The qRT-PCR results were obtained from three biological replicates.
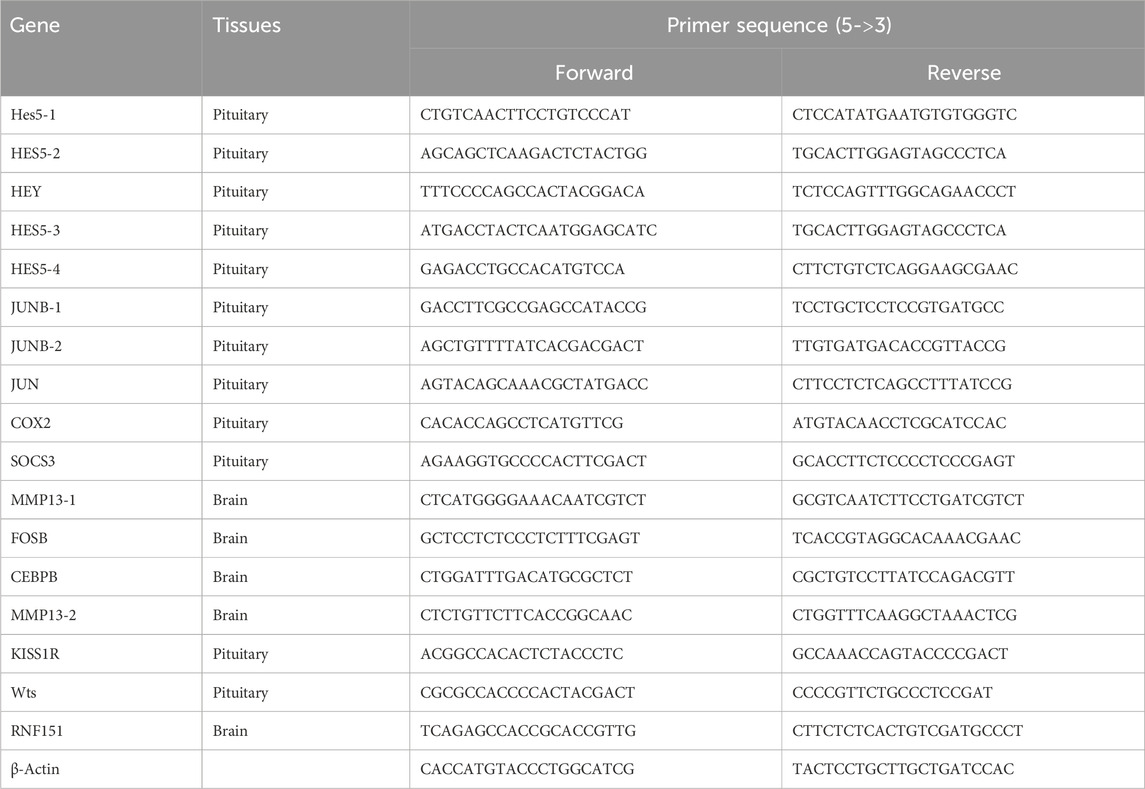
Table 1. Sequences of primers used for gene differential expression analysis.
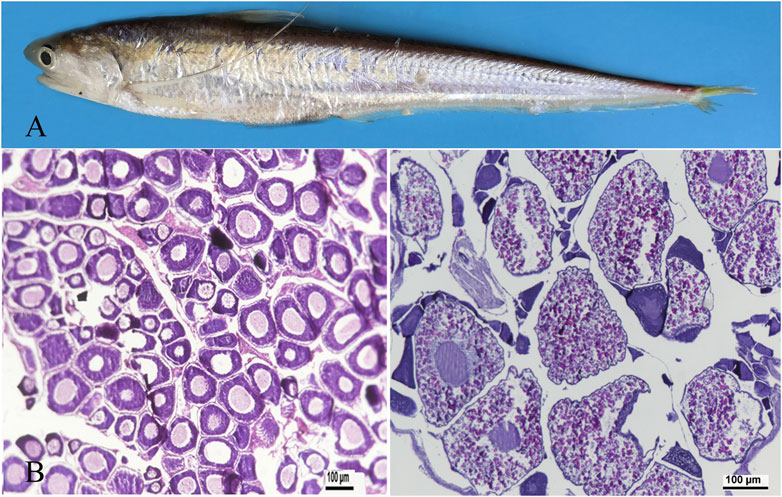
3 Results3.1 Histology of ovaryOvaries were isolated from adult and 3-year-old females (Figure 1A) and fixed in Bouin’s fixative solution. The fixed tissues were embedded in paraffin and sections were taken from ovaries and stained with haematoxylin. Images were captured using a Zeiss Axio Imager M2 microscope (Zeiss, Canada). At ovarian stage Ⅱ, primary oocytes enter early growth, and exhibit early, middle, and late phase oogonium. The cytoplasm is highly basophilic and dark blue. Oocytes are 80–120 μm in diameter (Figure 1B). At ovarian stage Ⅳ, the ovarian cells are eosinophilic, and the yolk is highly concentrated. The yolk particles are round, filled with the whole cell and reddish. The diameter of the ovarian cells is 200–250 μm (Figure 1C).
Figure 1. Representative photographs demonstrating ovarian developmental stages in adult taper-tail anchovy. (A) adult and female Coilia nasus. (B) ovary at stage Ⅱ. (C) ovary at stage Ⅳ. (Scale bar = 100 μm).
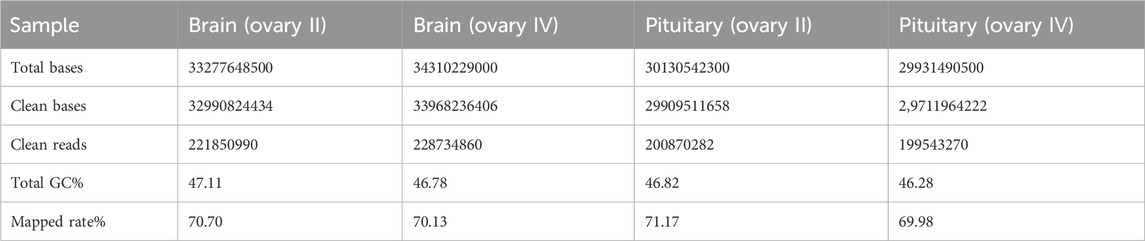
3.2 Sequencing of Coilia nasus transcriptomeTranscriptome sequencing was performed on the six C. nasus individuals using Illumina HiSeq high-throughput sequencing, including six brain libraries and six pituitary libraries. After quality control, a total of 126.48 Gb of clean data was generated, and the Q30 base percentage in each sample was at least 94.38%. The clean data from the six C. nasus individuals are summarized in Table 2.
Table 2. Statistical analysis of sequence quality for Coilia nasus transcriptome.
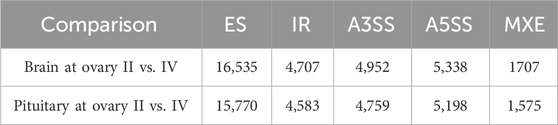
3.3 Alternative splicingThe alternative splicing events were analysed by rMATs software by comparing brain/pituitary at different stages of ovarian development using transcriptome assembly annotation (Supplementary Table S1), and five types of alternative splicing were detected, including exon skipping (ES), intron retention (IR), alternative 5′ splicing site (A5SS), alternative 3′ splicing site (A3SS) and mutually exclusive exon (MXE) (Table 3). Of these, seven alternative splicing transcripts were associated with sex-regulated and gonadal development (Table 4).
Table 3. Alternative splicing in brain/pituitary at different ovary stages.
Table 4. Alternative splicing of genes in regulating gonadal development.
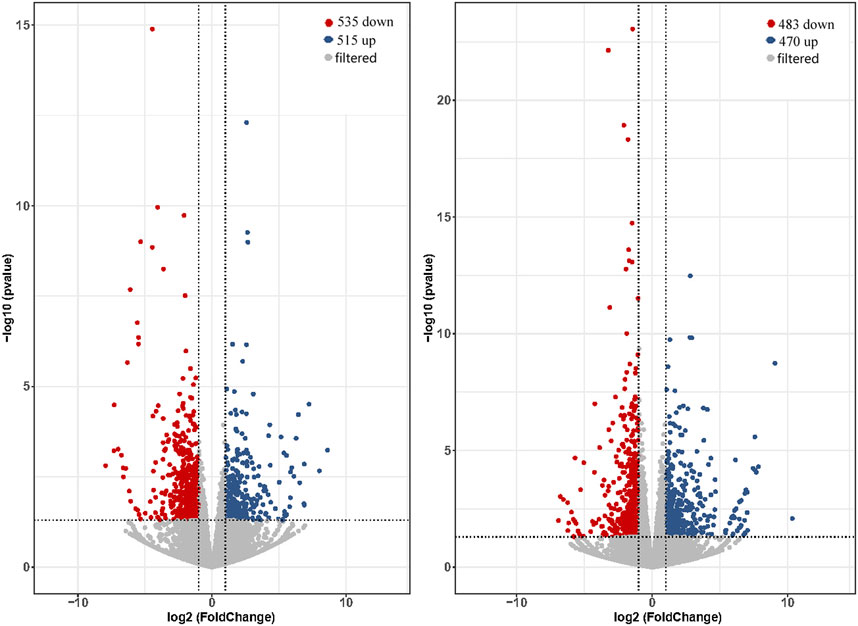
3.4 Analysis of differentially expressed genesThe expression levels of total genes were calculated by FPKM values and were 26.13%–30.1% in low expression (0.1–3.75 for FPKM), 24.18%–30.30% in medium expression (3.75–15 for FPKM) and 31.84%–34.64% in high expression (>15 for FPKM). Comparing of brain and pituitary with ovary at different developmental stages, the ovary-stage Ⅳ brain showed upregulation of 515 DEGs and downregulation of 535 DEGs compared to ovary-stage Ⅱ brain. On the other hand, ovary-stage Ⅳ pituitary showed upregulation of 470 DEGs and downregulation of 483 DEGs compared to ovary-stage Ⅳ pituitary. Volcano plots were shown in Figure 2 (p-value <0.05 and absolute value of log2 (fold change) > 2).
Figure 2. Volcano plot of DEGs calculated based on raw counts from brain (left) and pituitary (right) with ovarian development at stage Ⅳ versus stage Ⅱ.
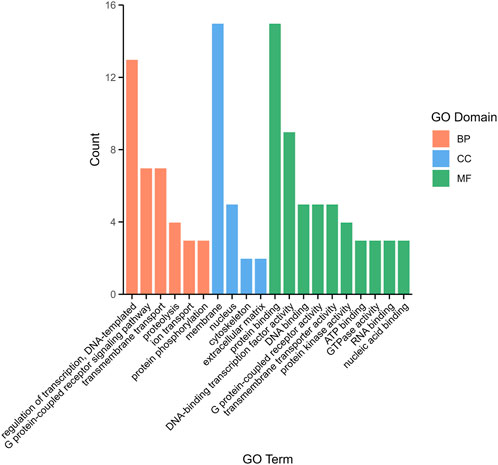
3.5 DEG annotation and enrichment analysisTo explore the function of DEGs, a total of 1,085 comparative combination DEGs were obtained from brain and pituitary with unsynchronized ovary and subjected to Gene Ontology (GO) analysis. Three categories were annotated as being associated with binding in terms of biological process (BP, 37.65%), cellular component (CC, 19.43%) and molecular function (MF, 46.96%). Among the BP, regulation of transcription, DNA-templated (5.26%), G protein-coupled receptor signaling pathway (2.83%) and transmembrane transport (2.83%) were the most highly represented GO terms, whereas in the CC, membrane (6.07%) and nucleus (2.02%), and in the MF, protein binding (6.07%) and DNA-binding transcription factor activity (3.64%) (Figure 3).
Figure 3. The top 20 distribution of DEGs among GO terms.
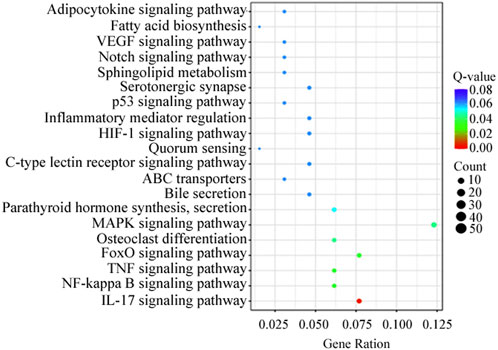
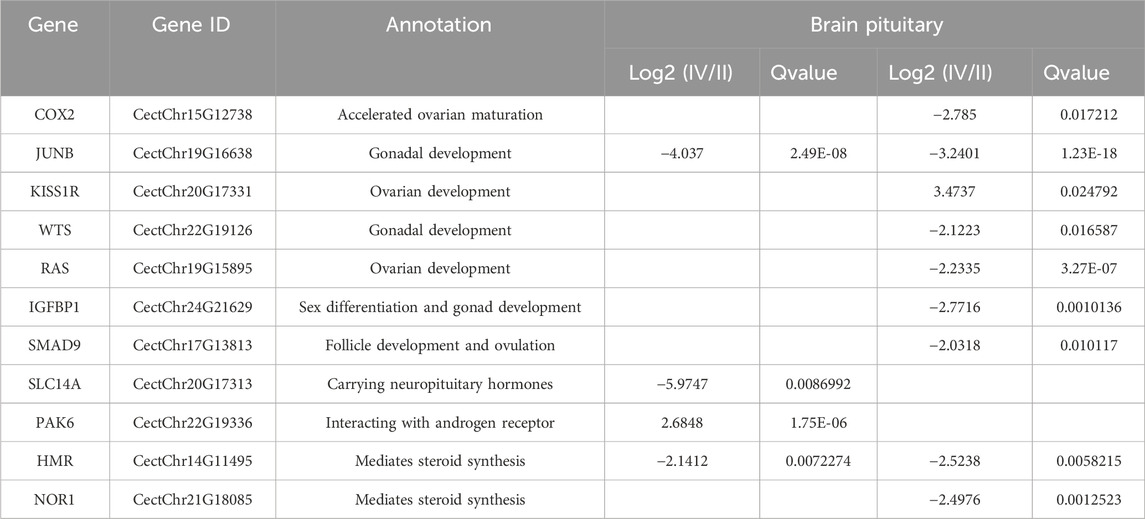
KEGG analysis revealed that the DEGs were significantly enriched in 103 KEGG pathways. The top 20 were enriched in biological processes, including MAPK signaling pathway (12.3%), IL-17 signaling pathway (7.7%) and FoxO signaling pathway (7.7%) (Figure 4). Genes present in the KEGG pathways involved in hormone synthesis and secretion pathway to regulate ovary development via hypothalamus-pituitary-gonadal axis (Table 5).
Figure 4. Top 20 distribution of DEGs presented in KEGG pathways.
Table 5. Gonad development-related DEGs in KEGG pathways.
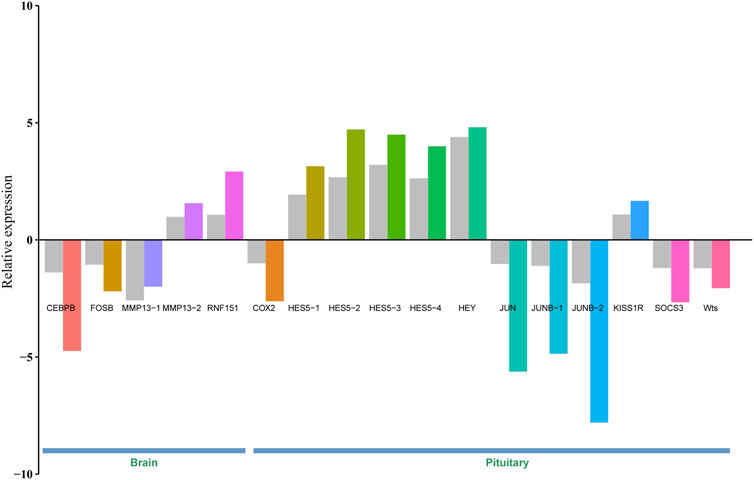
3.6 Validation of DEGsTo validate gene expression differences between brain/pituitary with ovary stage II and stage IV of C. nasus, 17 genes including CEBPB, FOSB, MMP13-1, MMP13-2, RNF151, COX2, HES5-1, HES5-2, HES5-3, HES5-4, HEY, JUN, JUNB-1 (jun B proto-oncogene 1), JUNB-2, KISS1R (Kiss1 receptor), SOCS3 and Wts were selected and applied a qRT-PCR method to determine their relative expression levels. Gene expression results from qRT-PCR were consistent with FPKM values from Illumina sequencing under the same conditions, indicating that DEGs from RNA-seq data were reliable (Figure 5).
Figure 5. Validation of 17 DEGs in the brain and pituitary with ovary at stage Ⅱ and Ⅳ by qRT-PCR. Grey bar denotes FPKM value calculated by eXpress, and colour bar denotes qRT-PCR value.
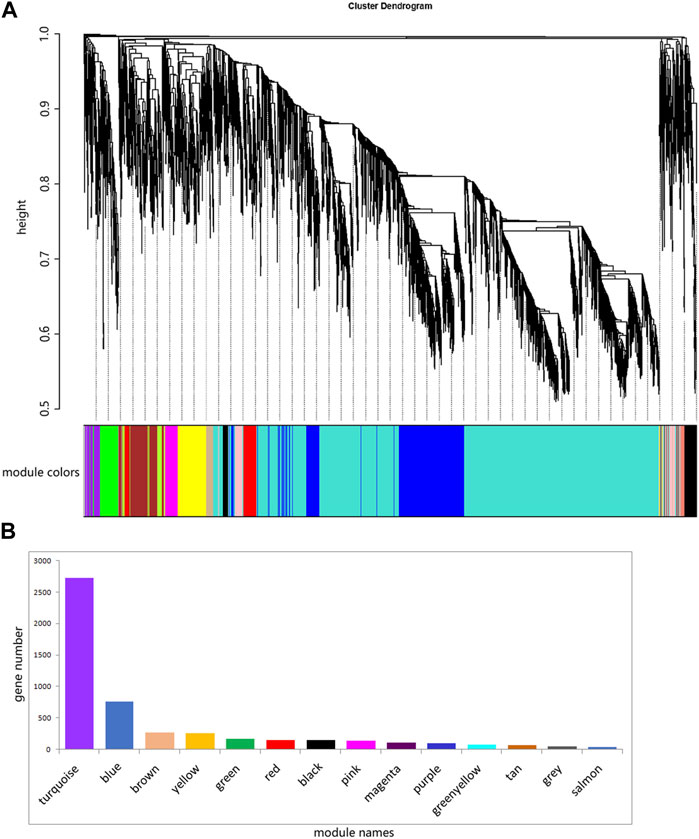
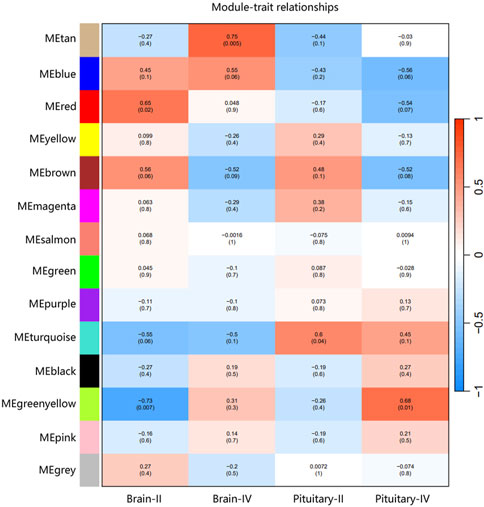
3.7 Weighted gene co-expression analysis (WGCNA) of expressed genesThe ovary development process of C. nasus is controlled by interacting gene networks, and the weighted gene co-expression network analysis (WGCNA) method was applied to identify the gene modules with similar expression profiles during the ovary development periods of C. nasus and to analyze their biological function and metabolic pathways. As a result, 14 modules were obtained after dynamic cutting (Figure 6A). These modules had distinct gene sizes, ranging from 2,728 in the turquoise module to 30 in the salmon module (Figure 6B). Correlation analyses were performed between modules and brain/pituitary with ovary phenotypic traits at different developmental stages (Figure 7). The “red” and “tan” modules were significantly positively correlated (r ≥ 0.6, p < 0.05) with brain where ovary developmental traits were at stage II and IV, respectively. The “turquoise” and “greenyellow” modules were significantly positively correlated (r ≥ 0.6, p < 0.05) with pituitary, where ovary developmental traits were at stage II and IV stage, respectively.
Figure 6. Fourteen different modules identified. (A) Gene co-expression network gene clustering number and modular cutting. The vertical distance of trees represents the distance between genes. Module colors are the module division of merged modules with similar expression patterns according to module similarity. (B) Number of genes per module. The abscissa represents each module, and the ordinate represents the number of genes.
Figure 7. Correlation between gene co-expression network modules and brain/pituitary with ovary different traits. The horizontal axis represents different traits, and the vertical axis represents each module. The colour lattice and number without bracket represent a correlation between the ovary traits with the module, while the number with bracket represents a significant degree of correlation.
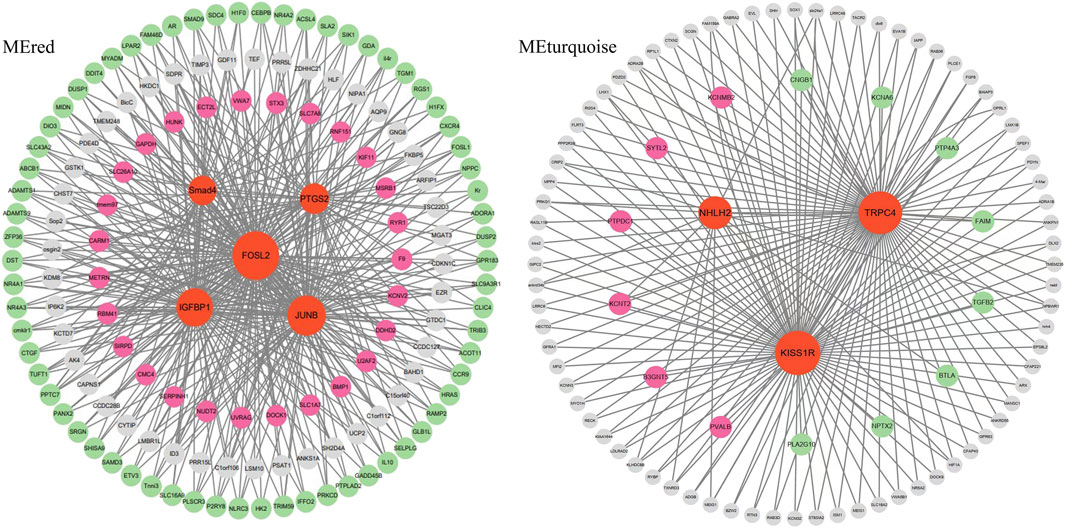
3.8 Identification of hub genes in ovary stage-related modulesHub genes were further identified in brain/pituitary modules with ovarian stage related traits. Genes with maximum abs (kME) value >0.9 and significance value <0.05 were selected as hub genes. A total of 47 (MEred), 12 (MEtan), 1,285 (ME turquoise) and 25 (ME greenyellow) hub genes were characterized in the four ovarian stage related modules (Figure 7). These modules were highly correlated with brain and pituitary with ovary stage traits (r ≥ 0.6). The key hub genes were screened out and evaluated based on the modules, including 1 gene regulating gonadotrophin secretion (Smad4, CectChr04G02957), 1 RNA-binding protein gene associated with brain developmental delay (R3HDM1, CectChr23G20501), 2 sensors of neurotransmission genes acting hypothalamic gonadotrophin-releasing hormone (TRPC4, CectChr17G13807; NHLH2, CectChr16G13558) and 1 gene regulating reproductive function (INSIG, CectChr06G04191).
Since Smad4, R3HDM1, TRPC4, NHLH2 and INSIG genes may play an important role in the brain-pituitary- gonadal axis, to further elucidate their functions in ovarian development, the co-expression gene networks of these genes were further analyzed. We found that Smad4 was co-expressed with DEGs including FOSL2, IGFBP1 and JUNB at the same time to form the main network. TRPC4, NHLH2 and KISS1R hub genes were co-expressed with either six up-expressed DEGs or eight down-expressed DEGs (Figure 8). R3HDM1 was co-expressed with two up- and six-expressed DEGs (Supplementary Figure S1). INSIG was co-expressed with 16 up- and 28 down-expressed DEGs (Supplementary Figure S1). These co-expressed hub genes were significantly enriched in signaling pathways related to ovarian development (Table 5), suggesting the reason for unsynchronized ovarian development leading to hormone release from the brain and pituitary.
Figure 8. The network construction of hub genes in brain and pituitary with ovary at different development stages in two modules, MEred and MEturquoise.
4 DiscussionTranscriptome analysis is an effective approach to uncover molecular mechanisms at the gene expression level. In our study, a brain/pituitary transcriptome analysis was performed to characterize the unsynchronized ovarian development of C. nasus and to uncover reproductive mechanisms by regulating the brain-pituitary-gonadal axis (Kanda, 2019). In most teleosts, ovarian development can be classified as synchronous or asynchronous according to the growth pattern of oocytes (Scott, 1987). In synchronous spawners, a mature egg can be produced as an annual event over the lifetime of the individual. In contrast, in asynchronous spawners, mature eggs are ovulated in multiple batches from a developing oocyte during each spawning season. Xu et al. report that C. nasus females spawn once a year, and individual ovary development is significantly different in stage (the ratio of stage III: stage IV: stage V was 3:3:4) within a spawning season, despite C. nasus being artificially cultured with consistent environmental conditions (Xu et al., 2011). Xue et al. applied ovarian transcriptomic and metabolomic analyses to discover key signaling pathways, such as steroid hormone biosynthesis, that influence ovarian development in C. nasus (Xu et al., 2016).
For the anadromous C. nasus, the ovarian transcriptome profiles revealed a large proportion of genes involved in energy production, amino acid transport and metabolism during spawning migration (Duan et al., 2015) via food intake for energy consumption and energy storage (Yin et al., 2020). Brain transcriptome analyses of C. nasus from the Yangtze River showed that the brain plays a more regulatory role during migration, and the signal transduction pathways and genes relevant to neurotransmitter receptors, hormones and KISS1R were upregulated, providing key regulatory genes for gonadal development of anadromous C. nasus (Wang et al., 2020).
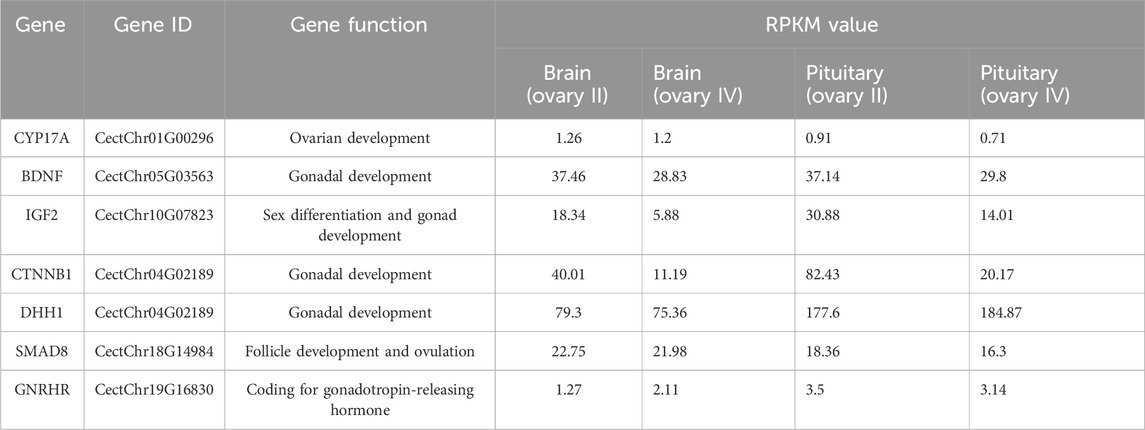
In our study, alternatively spliced transcript variants encoding different isoforms were found, and many gonad development-related transcript variants were detected in the C. nasus transcriptome data, including GnRHR (gonadotrophin-releasing hormone receptor), CYP17A1 (cytochrome P450 family 17 subfamily A 1), IGF-II (insulin-like growth factor II), and BDNF (brain-derived neurotrophic factor). GnRHR is mainly expressed in the pituitary gland and acts on the developing hypothalamic-pituitary-gonad axis by regulating sex hormone secretion (Li et al., 2022). CYP17A1 is a critical regulator of steroidogenesis in the conversion of pregnenolone to progesterone and, in the future, to 17-hydroxyprogesterone, which are processed to provide sex hormones. Mutant CYP17A1 resulted in abnormal sexual development (Singh et al., 2022). IGF-II is a reproduction-related growth factor, and can improve the developmental potential of oocytes, serving as a beneficial effect of growth factors in female reproduction (Muhammad et al., 2020). BDNF is heavily involved in the regulation of the hypothalamic-pituitary-adrenocortical axis (Naert et al., 2007). BDNF expression is correlated with gonadal hormones and its function is to regulate the products of gonadal steroids. Altered BDNF production and secretion has been implicated in a number of neurodegenerative diseases (Pluchino et al., 2013). These alternatively spliced transcripts obtained by comparing brain and pituitary at different stages of ovarian development suggested that alternatively spliced transcripts encoded by these genes may play an important role in reproduction of C. nasus.
Based on transcriptome data, DEGs affecting ovary growth were identified, including KISS1R, JUNB and IGFBP1. It was found that KISS1R, a key governor, provides the upstream signals for GnRH release, which subsequently regulates oocyte growth and maturation (Wang et al., 2011). Gonadotrophin hormones are glycoproteins produced by the pituitary gland that are required for normal reproductive function and gonadal development, and GnRH plays a fundamental role in the female reproductive system (Hollander-Cohen et al., 2021). Furthermore, KISS1R haploinsufficiency leads to premature ovarian failure, which is not rescued by ovarian gonadotrophin replacement (Gaytan et al., 2014). Interestingly, the kiss1 gene is absent in the gap-free genome of C. nasus (Gu et al., 2023), although JUNB showed significantly different expression in the brain and pituitary at different ovarian stages. JUNB was reported to be enriched within the Kiss1 promoter locus and showed a synergistic activation of Kiss1 promoter activity (Chakravarthi et al., 2018). IGFBP1 is a member of the soluble protein family, and either inhibits or stimulates the action of IGFs depending on the competition with IGF receptors for IGF-I, suggesting a potential role in the female reproductive ovary (Rutanen and Seppälä, 1992). The expression of the three genes was significantly different in brain and pituitary, and higher in pituitary with ovary at stage Ⅳ than Ⅱ for KISS1R, while lower in both brain and pituitary with ovary at stage Ⅳ than Ⅱ for JUNB and IGFBP1 in the ovarian development periods, and we speculate that the three genes have an important role in normal ovarian development.
WGCNA analysis of expressed genes identified 5 key hub genes from the “MEred”, “MEtan”, “MEturquoise” and “MEgreenyellow” modules. 1 gene regulating gonadotrophin secretion (Smad4), 1 brain developmental delay gene (R3HDM1), 2 genes acting hypothalamic gonadotrophin-releasing hormone (TRPC4, NHLH2) and 1 gene regulating reproductive function (INSIG) were important. These genes are involved in the brain-pituitary-gonadal axis, which is essential for ovarian development in fish. Ongaro et al. (Ongaro et al., 2020) reported that Smad4 stimulates the expression of follicle-stimulating hormone (FSH), an essential gonadotrophin hormone synthesized by pituitary gonadotrope cells. And the absence of Smad4 in gonadotropes renders females sterile (Fortin et al., 2014). The SMADs bind the FSH promoter in combination with the transcription factor Forkhead box L2, and an increase in FSH secretion is a key determinant of follicle maturation. In particular, there is an FSH threshold requirement for individual follicles, below which a given follicle will not develop (Odle and Childs, 2020). The hub genes in the “MEred” modules were more likely to be specific for ovarian development at stage Ⅳ, as four DEG hub genes (IGFBP1, FOSL2, JUNB and PTGS2) involved in fish reproduction were identified in these modules.
Meanwhile, the TRPC4 gene was a key candidate hub gene for ovarian development in C. nasus. TRPC4 is prominently expressed in the reproductive centre of the neuroendocrine brain, and TRPC4 activation contributes to kisspeptin to stimulate GnRH release (Götz et al., 2017). KISS1R is highly expressed in GnRH neurons and plays a crucial role in controlling pituitary gonadotrophin secretion and ultimately reproduction (Zhang et al., 2013). Network analysis of the hub gene TRPC4 found that the gene co-expressed with differentially expressed KISS1R gene were significantly function in governing GnRH neuronal excitability, which coincidentally regulate ovarian development in the reproductive axis in C. nasus, suggesting that they may co-regulate GnRH secretion in females, providing a plausible pathway for ovarian development at different stages.
5 ConclusionIn the present study, we detected 850/953 differentially expressed genes potentially associated with ovary development in the reproductive axis by transcriptome sequencing analysis of the brain/pituitary of C. nasus at different stages of ovary development. We identified 7 novel alternatively spliced transcripts, including CYP17A, IGF2 and GNRHR, that may affect ovarian development. In comparing the gene expression levels of brain/pituitary with ovary at differently developmental stages (stage Ⅳ vs stage Ⅱ) using RNA-seq, we identified 11 DEGS including KISS1R, IGFBP1 and WTS that may affect gonadotrophin secretion via regulation of the “hypothalamus-pituitary-gonad” axis. We also used WGCNA analysis to identify genes associated with gonadotrophin secretion in response to ovarian developmental traits. Four co-expression modules showed a high correlation with these ovarian stage traits, and the key hub genes Smad4, R3HDM1, TRPC4, NHLH2 and INSIG were screened out and their functions were involved in regulating ovarian tissue growth and development. Network analysis revealed that the key genes underlying these four modules were regulatory networks with DEGs. We provide a theoretical basis for delayed and unsynchronized ovarian development to improve artificial breeding of C. nasus.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statementThe animal study was approved by Animal Ethics Committee of Shanghai Ocean University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributionsZY: Data curation, Formal Analysis, Writing–original draft, Investigation. ZG: Formal Analysis, Investigation, Writing–original draft, Validation. YZe: Methodology, Writing–review and editing, Software, Visualization. ML: Writing–review and editing, Formal Analysis, Project administration, Resources. GX: Supervision, Project administration, Writing–review and editing. MR: Supervision, Writing–review and editing. YZh: Conceptualization, Data curation, Funding acquisition, Project administration, Writing–review and editing. DL: Conceptualization, Funding acquisition, Methodology, Resources, Validation, Writing–original draft, Writing–review and editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China (2022YFD2400904).
AcknowledgmentsWe are grateful to all workers at the experimental sites.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2024.1361386/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The network construction of hub genes in brain and pituitary with ovary at different development stages in MEtan and MEgreenyellow modules.
SUPPLEMENTARY TABLE S1 | Transcriptome annotation for brain and pituitary of Coilia nasus.
ReferencesAmberg, J. J., Goforth, R. R., and Sepúlveda, M. S. (2013). Antagonists to the Wnt cascade exhibit sex-specific expression in gonads of sexually mature shovelnose sturgeon. Sex. Dev. 7, 308–315. doi:10.1159/000354280
PubMed Abstract | CrossRef Full Text | Google Scholar
Bókony, V., Milne, G., Pipoly, I., Székely, T., and Liker, A. (2019). Sex ratios and bimaturism differ between temperature-dependent and genetic sex-determination systems in reptiles. BMC Evol. Biol. 19, 57. doi:10.1186/s12862-019-1386-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Cao, H., Gao, H., Li, Z., Peng, G., Chen, Y., Jin, T., et al. (2022). Comparative transcriptome provides insights into differentially expressed genes between testis and ovary of Onychostoma macrolepis in reproduction period. Gen. Comp. Endocrinol. 326, 114066. doi:10.1016/j.ygcen.2022.114066
PubMed Abstract | CrossRef Full Text | Google Scholar
Chakravarthi, V. P., Khristi, V., Ghosh, S., Yerrathota, S., Dai, E., Roby, K. F., et al. (2018). ESR2 is essential for gonadotropin-induced Kiss1 expression in granulosa cells. Endocrinology 159, 3860–3873. doi:10.1210/en.2018-00608
PubMed Abstract | CrossRef Full Text | Google Scholar
Dechaud, C., Miyake, S., Martinez-Bengochea, A., Schartl, M., Volff, J. N., and Naville, M. (2021). Clustering of sex-biased genes and transposable elements in the genome of the medaka fish oryzias latipes. Genome Biol. Evol. 13, evab230. doi:10.1093/gbe/evab230
PubMed Abstract | CrossRef Full Text | Google Scholar
Dong, J., Li, J., Hu, J., Sun, C., Tian, Y., Li, W., et al. (2020). Comparative genomics studies on the dmrt gene family in fish. Front. Genet. 11, 563947. doi:10.3389/fgene.2020.563947
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, F. K., Xu, G. C., Nie, Z. J., Xu, P., and Gu, R. B. (2014). Transcriptome analysis gene expression in the liver of Coilia nasus during the stress response. BMC Genomics 15, 558. doi:10.1186/1471-2164-15-558
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, X., Wang, B., Liu, X., Liu, X., He, Y., Zhang, Q., et al. (2017). Comparative transcriptome analysis of ovary and testis reveals potential sex-related genes and pathways in spotted knifejaw Oplegnathus punctatus. Gene 637, 203–210. doi:10.1016/j.gene.2017.09.055
PubMed Abstract | CrossRef Full Text | Google Scholar
Duan, J. R., Zhou, Y. F., Xu, D. P., Zhang, M. Y., Liu, K., Shi, Y., et al. (2015). Ovary transcriptome profiling of Coilia nasus during spawning migration stages by Illumina sequencing. Mar. Genomics 21, 17–19. doi:10.1016/j.margen.2015.02.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Farhadi, A., Fang, S., Zhang, Y., Cui, W., Fang, H., Ikhwanuddin, M., et al. (2021). The significant sex-biased expression pattern of Sp-Wnt4 provides novel insights into the ovarian development of mud crab (Scylla Paramamosain). Int. J. Biol. Macromol. 183, 490–501. doi:10.1016/j.ijbiomac.2021.04.186
PubMed Abstract | CrossRef Full Text | Google Scholar
Fortin, J., Boehm, U., Deng, C. X., Treier, M., and Bernard, D. J. (2014). Follicle-stimulating hormone synthesis and fertility depend on SMAD4 and FOXL2. Faseb J. 28, 3396–3410. doi:10.1096/fj.14-249532
PubMed Abstract | CrossRef Full Text | Google Scholar
Gaytan, F., Garcia-Galiano, D., Dorfman, M. D., Manfredi-Lozano, M., Castellano, J. M., Dissen, G. A., et al. (2014). Kisspeptin receptor haplo-insufficiency causes premature ovarian failure despite preserved gonadotropin secretion. Endocrinology 155, 3088–3097. doi:10.1210/en.2014-1110
PubMed Abstract | CrossRef Full Text | Google Scholar
Götz, V., Qiao, S., Beck, A., and Boehm, U. (2017). Transient receptor potential (TRP) channel function in the reproductive axis. Cell Calcium 67, 138–147. doi:10.1016/j.ceca.2017.04.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Gu, K., Zhang, Y., Zhong, Y., Kan, Y., Jawad, M., Gui, L., et al. (2023). Establishment of a Coilia nasus spermatogonial stem cell line capable of spermatogenesis in vitro. Biol. (Basel) 12, 1175. doi:10.3390/biology12091175
CrossRef Full Text | Google Scholar
Hayes, T. B. (1998). Sex determination and primary sex differentiation in amphibians: genetic and developmental mechanisms. J. Exp. Zool. 281, 373–399. doi:10.1002/(SICI)1097-010X(19980801)281:5<373::AID-JEZ4>3.0.CO;2-L
PubMed Abstract | CrossRef Full Text | Google Scholar
Hollander-Cohen, L., Golan, M., and Levavi-Sivan, B. (2021). Differential regulation of gonadotropins as revealed by transcriptomes of distinct LH and FSH cells of fish pituitary. Int. J. Mol. Sci. 22, 6478. doi:10.3390/ijms22126478
PubMed Abstract | CrossRef Full Text | Google Scholar
Kaitetzidou, E., Gilfillan, G. D., Antonopoulou, E., and Sarropoulou, E. (2022). Sex-biased dynamics of three-spined stickleback (Gasterosteus aculeatus) gene expression patterns. Genomics 114, 266–277. doi:10.1016/j.ygeno.2021.12.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Kan, Y., Zhong, Y., Jawad, M., Chen, X., Liu, D., Ren, M., et al. (2022). Establishment of a Coilia nasus gonadal somatic cell line capable of sperm induction in vitro. Biol. (Basel) 11, 1049. doi:10.3390/biology11071049
CrossRef Full Text | Google Scholar
Kanda, S. (2019). Evolution of the regulatory mechanisms for the hypothalamic-pituitary-gonadal axis in vertebrates-hypothesis from a comparative view. Gen. Comp. Endocrinol. 284, 113075. doi:10.1016/j.ygcen.2018.11.014
PubMed Abstract | CrossRef Full Text | Google Scholar
Li, X., Zhang, X., Shen, Z., Chen, Z., Wang, H., and Zhang, X. (2022). GnRH receptor mediates lipid storage in female adipocytes via AMPK pathway. Int. J. Med. Sci. 19, 1442–1450. doi:10.7150/ijms.74335
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, S., Han, C., and Zhang, Y. (2023). De novo assembly, characterization and comparative transcriptome analysis of gonads reveals sex-biased genes in Coreoperca whiteheadi. Comp. Biochem. Physiol. Part D. Genomics Proteomics 47, 101115. doi:10.1016/j.cbd.2023.101115
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, X., Dai, S., Wu, J., Wei, X., Zhou, X., Chen, M., et al. (2022). Roles of anti-Müllerian hormone and its duplicates in sex determination and germ cell proliferation of Nile tilapia. Genetics 220, iyab237. doi:10.1093/genetics/iyab237
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, X., Ji, K., Jo, A., Moon, H. B., and Choi, K. (2013). Effects of TDCPP or TPP on gene transcriptions and hormones of HPG axis, and their consequences on reproduction in adult zebrafish (Danio rerio). Aquat. Toxicol. 134–135, 104–111. doi:10.1016/j.aquatox.2013.03.013
PubMed Abstract | CrossRef Full Text | Google Scholar
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi:10.1006/meth.2001.1262
PubMed Abstract | CrossRef Full Text | Google Scholar
Ma, F., Wang, Y., Su, B., Zhao, C., Yin, D., Chen, C., et al. (2023). Gap-free genome assembly of anadromous Coilia nasus. Sci. Data 10, 360. doi:10.1038/s41597-023-02278-w
PubMed Abstract | CrossRef Full Text | Google Scholar
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi:10.1038/nmeth.1226
PubMed Abstract | CrossRef Full Text | Google Scholar
Muhammad, T., Li, M., Wang, J., Huang, T., Zhao, S., Zhao, H., et al. (2020). Roles of insulin-like growth factor II in regulating female reproductive physiology. Sci. China Life Sci. 63, 849–865. doi:10.1007/s11427-019-1646-y
PubMed Abstract | CrossRef Full Text | Google Scholar
Naert, G., Maurice, T., Tapia-Arancibia, L., and Givalois, L. (2007). Neuroactive steroids modulate HPA axis activity and cerebral brain-derived neurotrophic factor (BDNF) protein levels in adult male rats. Psychoneuroendocrinology 32, 1062–1078. doi:10.1016/j.psyneuen.2007.09.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Ongaro, L., Schang, G., Zhou, Z., Kumar, T. R., Treier, M., Deng, C. X., et al. (2020). Human follicle-stimulating hormone ß subunit expression depends on FOXL2 and SMAD4. Endocrinology 161, bqaa045. doi:10.1210/endocr/bqaa045
PubMed Abstract | CrossRef Full Text | Google Scholar
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi:10.1038/nbt.3122
PubMed Abstract | CrossRef Full Text | Google Scholar
Pluchino, N., Russo, M., Santoro, A. N., Litta, P., Cela, V., and Genazzani, A. R. (2013). Steroid hormones and BDNF. Neuroscience 239, 271–279. doi:10.1016/j.neuroscience.2013.01.025
PubMed Abstract | CrossRef Full Text | Google Scholar
Rutanen, E. M., and Seppälä, M. (1992). Insulin-like growth factor binding protein-1 in female reproductive functions. Int. J. Gynaecol. Obstet. 39, 3–9. doi:10.1016/0020-7292(92)90772-b
PubMed Abstract | CrossRef Full Text | Google Scholar
Schartl, M., Wilde, B., Schlupp, I., and Parzefall, J. (1995). Evolutionary origin of a parthenoform, the amazon molly poecilia formosa, on the basis of a molecular genealogy. Evolution 49, 827–835. doi:10.1111/j.1558-5646.1995.tb02319.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Scott, A. P. (1987). “Reproductive endocrinology of fish,” in Fundamentals of comparative vertebrate endocrinology. Editors I. Chester-Jones, P. M. Ingleton, and J. G. Phillips (Boston: Springer Press), 223–256.
留言 (0)