MTX-resistant cell line establishment
The original HT29 cell line (S-0, stage 0) was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China, http://www.cellbank.org.cn/). Initially, this cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM; GibcoBRL, Gaithersburg, MD, USA) supplemented with 1.0 × 10−7 mol/L MTX (Pfizer (Perth) Pty, Bentley WA, Australia) to induce resistance. After cell growth was stable, the next concentration of MTX was added successively to induce drug resistance until the concentration of MTX was 6.0 × 10−4 mol/L. All the cell lines (S-1-S-18:1.0 × 10−7, 2.0 × 10−7, 4.0 × 10−7, 6.0 × 10−7, 8.0 × 10−7, 1.0 × 10−6, 2.0 × 10−6, 4.0 × 10−6, 6.0 × 10−6, 8.0 × 10−6, 1.0 × 10−5, 2.0 × 10−5, 4.0 × 10−5, 6.0 × 10−5, 8.0 × 10−5, 1.0 × 10−4, 3.0 × 10−4 and 6.0 × 10−4 mol/L of MTX resistant cells) were cultured in the presence of 15% fetal calf serum (PAA Laboratories GmbH, Pasching, Austria). Human osteosarcoma cell line U2OS (Shanghai, China, http://www.cellbank.org.cn/) and its MTX-resistant cell lines U2OS e-6 and U2OS e-4 (the concentrations of MTX resistance were 1.0 × 10−6 mol/L and 1.0 × 10−4 mol/L, respectively), previously constructed by our research group, were also cultured in DMEM with 15% fetal calf serum. All cell lines were negative for Mycoplasma contamination.
Characterization of the MTX-resistant cell lines
Cell viability was assessed using CellTiter 96® AQueous One Solution (Promega, Madison, WI, USA). Cells (5000/well) were treated with different concentrations of MTX from 1.0 × 10−9 mol/L to 1.0 × 10−2 mol/L for 72 h and incubated with 20 μl 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) for 3 h. Then optical density (OD) values of the MTS solution were measured using a microplate reader (Tecan, Grödig, Austria) at 492 nm wavelength. The values of the half-maximal inhibitory concentration (IC50) were calculated.
DHFR gene copy numbers were measured using real-time PCR performed with the LightCycler 480 system (Roche Applied Science, Mannheim, Germany). Genomic DNA was extracted using a QIAmp DNA Mini Kit (Qiagen, Düsseldorf, Germany) following the instructions from the manufacturer. The DNA primers used are listed in Table S10 with β-actin as control. The amplification steps of all these genes were performed for 45 cycles of 20 s at 95 °C, 20 s at 60 °C and 30 s at 72 °C.
Metaphase spreads from Colcemid (Sigma-Aldrich Co.LLC, Saint-Louis, MO, USA) arrested cells were prepared according to standard cytogenetic methods [15]. DNA from the BAC clones PR11-90A9 (BAC PAC Resources Center, Oakland, CA, USA) and/or RP11-91I22 (BAC PAC Resources Center, Oakland, USA) was extracted using a Genopure Plasmid Midi Kit (Roche Applied Science, Mannheim, Germany) and labeled with Green-dUTP and Red-dUTP, respectively, using the BioPrime DNA Labeling System Kit (Invitrogen, Carlsbad, CA, USA). Hybridization to metaphase spreads was as described in our previous study [15], and the slides were counterstained with DAPI. Images were obtained using a fluorescence microscope equipped with the MetaMorph Imaging System 7.7.0.0 (Molecular Devices Corporation, Sunnyvale, CA, USA).
Genome-wide CNV analyses
Genomic DNA (>1.5 μg) was randomly segmented to an average of 350 bp and subjected to DNA library creation with the Illumina NGS DNA Library Construction Kit (Illumina, San Diego, CA, USA). Whole-genome sequencing data were generated by Novogene (Beijing, China) using PCR-based libraries and 150 base paired-end sequencing on the Illumina HiSeqX Ten platform. The mean sequence depth on the 19 cell lines was 32×, with mean coverage of the genome 98.94% (Table S2). Control-FREEC was applied to call CNVs from NGS data. Copy numbers of chromosomal segments are shown in Supplementary Material 1. A SNP array was also used to detect copy number changes. CNV calling from SNP array data (Figs. S1, S2 and Table S8,S9), and the comparison of CNVs from sequencing and SNP genotyping data are shown in Table S3. We counted the total amount of amplified (gained) DNA by adding together the length of all CNVs with copy number greater than 2, and the total amount of deleted (lost) DNA by adding the length of all CNVs with copy number less than 2 in each cell line. The net amount of DNA changes was calculated as the total gain minus the total loss. Lowess curve fitting was performed on the total DNA gain, loss and net change plotted against MTX-resistance level, and the Pearson correlation was calculated. All these analyses were performed using R (R-3.2.0, win-64).
MTX-resistance-specific CNV analyses
CNVs called from sequencing data were used for this analysis. We identified the CNVs in each of the MTX-resistant HT29 cell lines (S1−S18) which differed from the original HT29 cell line (S0), requiring less than 50% overlap between them. We defined this set of CNVs as MTX-resistance-specific CNVs (MRS-CNVs). We also filtered out MRS-CNVs smaller than 10 kb (an arbitrary choice) (Figure S3).
As both genome-wide CNVs and MRS-CNVs were called per sample, we first merged all MRS-CNVs into a union call set containing the calls from all of the 18 cell lines. We kept cell-line-specific MRS-CNVs and shared ones with the same start and end coordinates as they were, but split the partially shared ones into cell-line-specific and completely shared ones. We then assigned genotypes for all of the union set across all of the 18 MTX-resistant cell lines. We finally weighted these CNVs (length of basic CNV × copy number of the basic CNV/ total length of all basic CNVs) for the hierarchical clustering and principal component analyses. Both analyses were performed with R (R-3.2.0, win-64).
Genes that completely overlapped with MRS-CNVs were obtained using GENCODE 19 annotation on Ensembl GRCh37. These genes were further annotated as contributing to MTX-resistance or not, using the information from the Pharmacogenomics Knowledgebase (PharmGKB, https://www.pharmgkb.org/index.jsp) database.
The copy number and mRNA expression estimates for DHFR, MSH3, ZFYVE16, FAM151B, ANKRD34B, SPZ1 and MTRNR2L2 were validated using real-time PCR with the primers listed in Table S10, using β-actin as control. The PCR cycles were 20 s at 95 °C, 20 s at 60 °C and 30 s at 72 °C for 45 cycles. The protein expression level was measured using Western Blot with Beta Actin, Alpha Tubulin or GAPDH for normalization. The antibodies used and their sources are listed in Table S11.
siRNA transfection
Cells were seeded into 6-well plates at a density of 2 × 104 cells per well. Transfection was performed using 80 pmol of small interfering RNAs (siRNAs) (siDHFR, siMSH3, siZFYVE16 and siFAM151B) or control siRNA. jetPRIME reagent (PolyPlus Transfection, Strasbourg, France) was used for transfection following the manufacturer’s instructions. Transfection efficiency was assessed after 48 h. The sequences of siRNAs are listed in Table S12.
Cell counting kit-8 (CCK-8) assay
To analyze the effects of DHFR, MSH3, ZFYVE16 and FAM151B on MTX resistance, cells transfected with target gene siRNAs and control siRNA were harvested and seeded into 96-well plates at a density of 3000 cells per well. The cells were then treated with MTX for 48 h. Following this incubation period, the reagent of CCK-8 (GLPBIO, Montclair, CA, USA) was added to each well, and the plates were incubated for 2 h. Absorbance at 450 nm was measured to calculate the IC50 value for MTX.
Statistics
Statistical analysis was performed using the R program (v3.2.3). Differences between different groups (sample size = 3) were analyzed using a two-tailed Student′s t-test or one-way ANOVA analysis. The correlation coefficient was calculated using the Pearson method with R built-in function “cor()”. A Lowess curve was fitted with the R built-in function “lowess()”. Measurement data were presented as mean ± standard deviation (SD) of three independent experiments. Prior to the analysis, a normality test and a homogeneity of variance test were performed on all collected data. Significance was indicated by asterisks: *P < 0.05; **P < 0.01; ***P < 0.001.



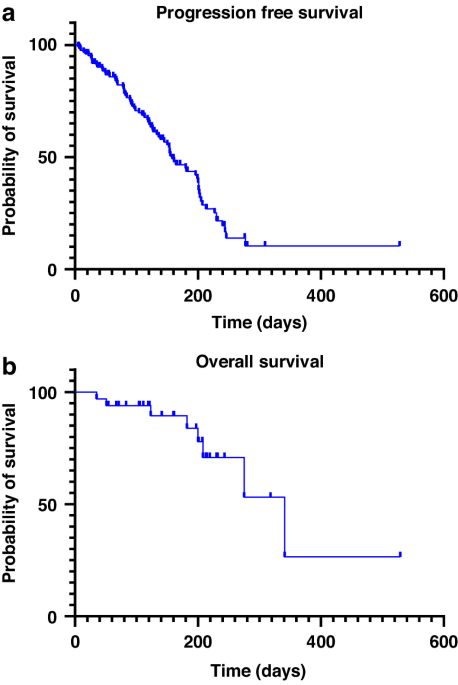
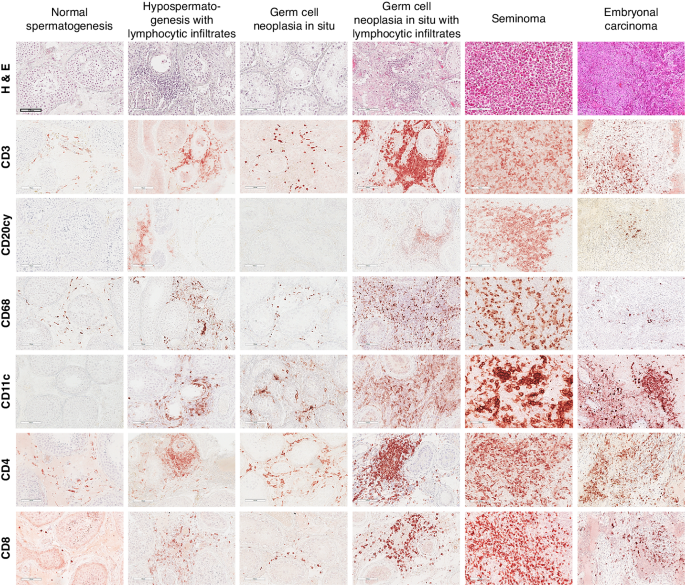
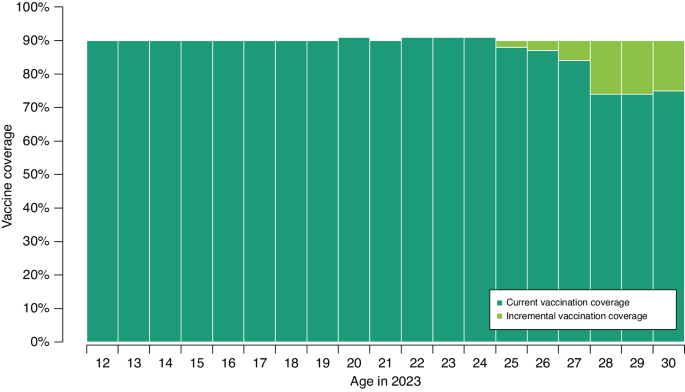
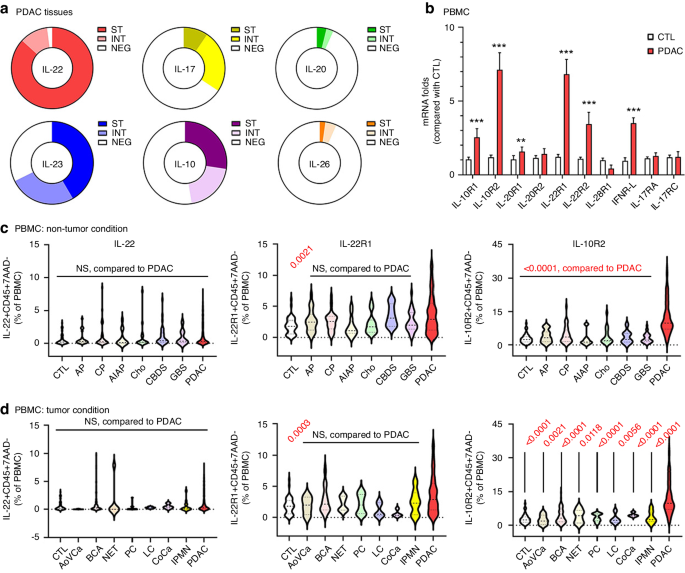

留言 (0)