
記住我




Williams syndrome (WS) is a distinctive multisystem disorder that affects the connective tissues and the cardiovascular and central nervous systems, and it is commonly associated with behavioral, developmental, and cardiovascular abnormalities. WS is caused by a de novo deletion of 1.5–1.8 Mb of 26–28 genes located on chromosome 7 at position 7q11.23 (1), with a prevalence rate of 1 in 8,000–20,000 newborns (2, 3). Although cardiovascular defects occur in approximately 80% of patients with WS (4), only 33% of diagnosed cardiovascular defects are detected prenatally, and they tend to be mild, so they are often ignored before birth (5). Structural cardiovascular abnormalities are present in up to 93% of patients with WS and present within the first year of life, with peripheral pulmonary artery stenosis and supravalvular aortic stenosis (SVAS) being the most common (6).
Endocardial calcification is a non-specific reaction to severe etiologically heterogeneous myocardial injuries (7). The etiology of endocardial calcification can be classified into two categories: dystrophic and metastatic, with dystrophic calcification being the main cause. Endocardial calcification may be related to hypoxic-ischemic injury; trauma, such as cardiac surgery; myocarditis (usually fungal or viral); or toxic damage due to alcohol or drugs consumption. Metastatic calcification may be present in patients with hyperparathyroidism, chronic renal failure, oxaluria and aluminum intoxication, dietary calcium and/or vitamin D deficiency, and sarcoidosis (7).
Endocardial calcification is uncommon during infancy. In this report, we present a case of a child with Williams syndrome (WS) who exhibited a typical cardiac structural defect along with rapid and progressive endocardial calcification within the left ventricle (LV).
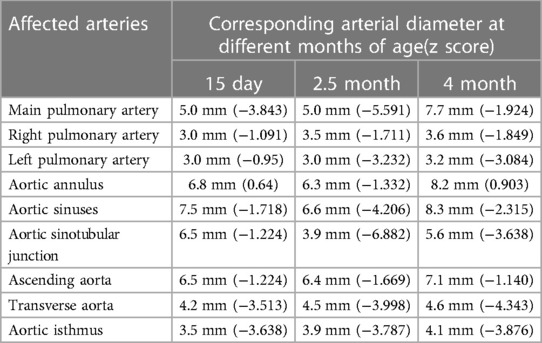
2 Case presentationA newborn baby was admitted to the neonatal department due to severe pneumonia, and she was noted to have a murmur. The baby was born to a 28-year-old G1P0 mother after elective induction at 36 + 1 weeks of gestation due to fetal distress. The neonatal birth weight was 2,100 g (p10), and the length was 44 cm (p15). The prenatal examination showed no abnormalities, except for intrauterine growth retardation. The family history was non-contributory, and the mother was not exposed to radiation or toxic substances during pregnancy. An echocardiogram at 15 days (Table 1) revealed mild-to-moderate pulmonary artery stenosis (Vmax = 3.0–3.5 m/s), and extremely mild aortic arch stenosis with a gradient of 20 mmHg with normal LV function (ejection fraction [EF] = 62%, lateral E/e′ [E/e′ Lat] = 7.8).
Table 1. The diameter of the affected arteries at different ages in a patient with Williams syndrome measured by echocardiography.
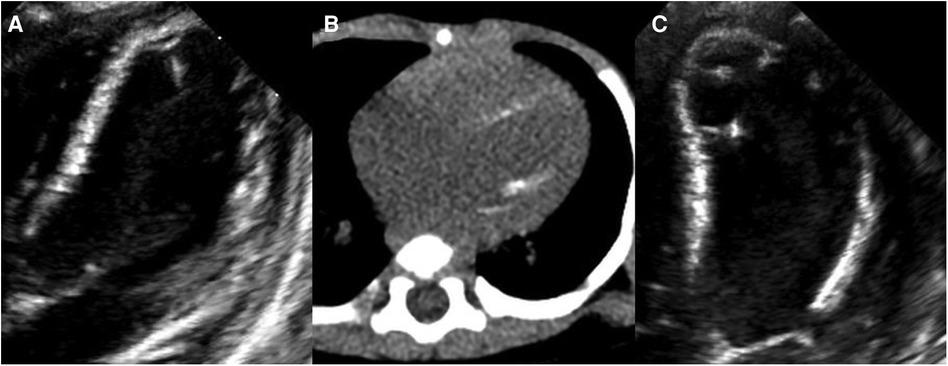
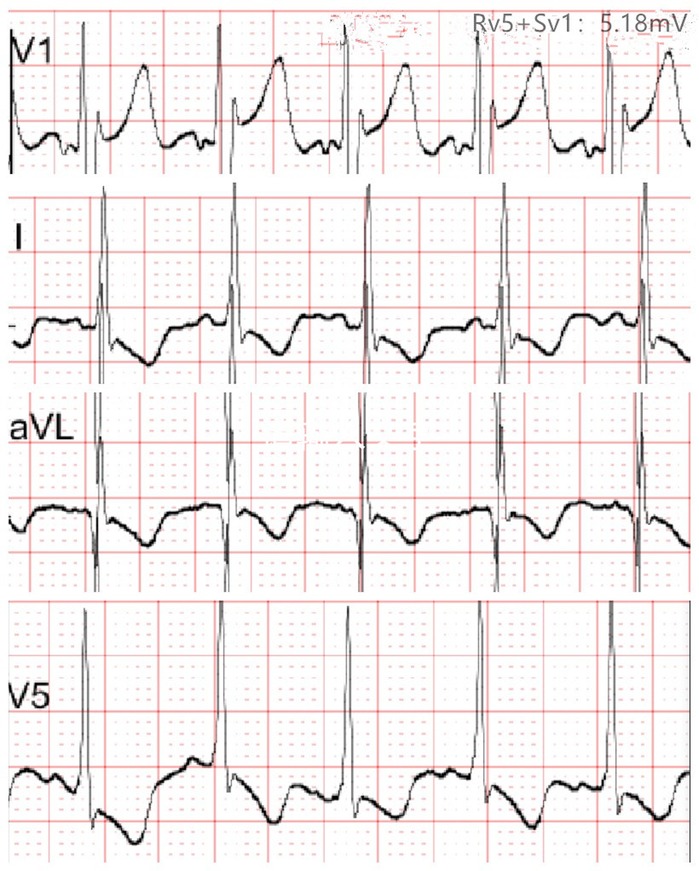
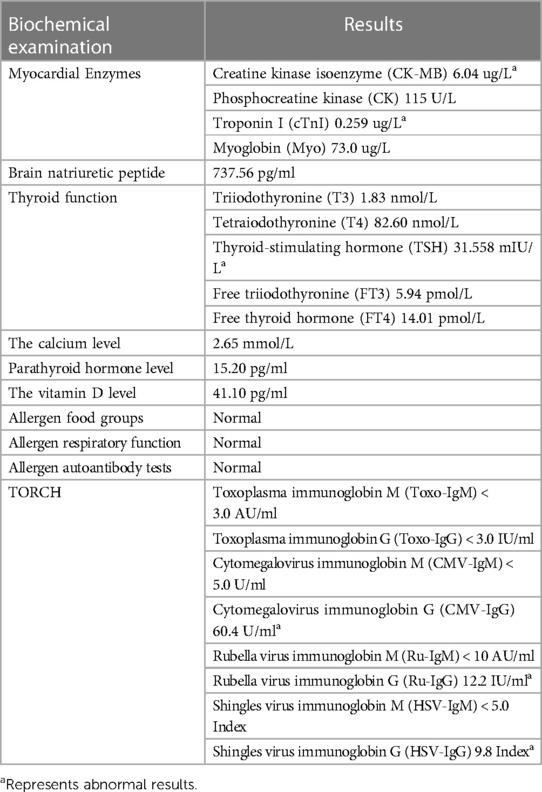
At 2.5 months of age, the patient was admitted to our hospital due to difficulty with feeding for more than 2 months, which was initially thought to be a result of her severe reflux. The echocardiogram (Table 1) showed moderate-to-severe stenosis of the main pulmonary artery and its branches (Vmax = 3.3–4.2 m/s), a gradient of 55 mmHg across the upper aortic valve, mild aortic arch stenosis with a gradient of 36 mmHg in the setting of LV dysfunction (EF = 35%, E/e′ Lat = 16.2), and increased echo of LV endocardium (Figure 1A). The coronary arteries appeared normal. Computed tomography angiography showed aortic stenosis, pulmonary artery stenosis, and diffusely distributed calcification of the LV endocardium (Figure 1B). The patient's eyelids were swollen, and blood pressure was normal (92/32 mmHg). The electrocardiogram (Figure 2) showed ST-T changes, and LV hypertrophy was suspected. Biochemical examination (Table 2) showed myocardial injury; subclinical hypothyroidism; TORCH complex infection; normal hepatic and renal function; normal calcium (2.65 mmol/L), parathyroid hormone, and vitamin D; and no abnormalities in allergen food groups, respiratory function, and autoantibody tests. WS was highly suspected based on these findings.
Figure 1. Left ventricular (LV) calcification detected by echocardiography and computed tomography angiography (CTA). At 2.5 months of age, (A) echocardiography showing increased echo of LV endocardium and (B) CTA showing diffusely distributed calcification of the LV endocardium. At 4 months of age, (C) echocardiography showing diffusely distributed calcification of the LV endocardium, chordae tendineae, and papillary muscle.
Figure 2. Electrocardiogram at 2.5 months of age showing T-wave flattening or inversion in leads I, aVL, and V5, and left ventricular hypertrophy was suspected.
Table 2. Biochemical examination indicators at 2.5 months of age in a patient with Williams syndrome.
Whole-exome sequencing of peripheral blood from the baby and her parents revealed a new pathogenic copy number variation (1.431Mb deletion) in the 7q11.23 region (Chr7: 73303398–74733981), which contains ELN, confirming the diagnosis of WS. The baby was given symptomatic treatment, including prednisolone acetate, digoxin as a cardiotonic glycoside, and captopril for diuresis. Echocardiography at the age of 4 months showed improved LV systolic function (EF = 42%, E/e′ Lat = 15.9) with diffusely distributed calcification of the LV endocardium, chordae tendineae, and papillary muscle (Figure 1C). Main pulmonary artery stenosis and SVAS had improved in lesion severity, while peripheral pulmonary artery stenosis and coarctation were unchanged (Table 1).
3 DiscussionWS is a rare disease that can affect multiple systems, with cardiovascular defects being the most common cause of death (8). Cardiovascular abnormalities are the direct result of ELN deletion (5). In patients with WS, the decrease in arterial elastin content and pathological alignment of elastin fibers lead to smooth muscle overgrowth, multilayer thickening of the media of large arteries, and development of obstructive hyperplastic intimal lesions, leading to vascular diseases in the middle- and large-sized arteries. The most notable stenoses are SVAS and peripheral pulmonary artery stenosis; however, the aortic arch, descending aorta, coronary artery, renal artery, mesenteric artery, and intracranial artery can also be affected (9). Other cardiovascular defects, including tetralogy of Fallot, complete atrioventricular septal defect, total anomalous pulmonary venous return, double-chambered right ventricular, and Ebstein anomaly of the tricuspid valve, have also been reported (10). In the present case, the patient had ELN deletion and typical pulmonary artery stenosis, SVAS, and aortic arch stenosis.
Although the typical defect of arterial stenosis and related clinical tests led us to quickly make a diagnosis of WS, which was confirmed by whole-exome sequencing, the etiology of the infant's rapidly progressive endocardial calcification was still unclear. Endocardial calcification is very rare in infancy. At present, only one case of giant cell myocarditis and endomyocardial calcification in a 2.5-month-old infant has been reported, which was triggered by excessive maternal alcohol abuse (11). The patient had poor cardiac function, and electrocardiogram indicated myocardial ischemia. Although the combination of high end-diastolic pressure due to severe SVAS coupled with coronary artery stenosis can result in decreased coronary artery perfusion pressure and myocardial ischemia (12), ultimately resulting in cardiac dysfunction and papillary muscle and endocardial calcification, it often takes a long time to develop. In the present study, echocardiography showed no significant coronary artery stenosis, so the decrease in cardiac function and endocardial calcification could not be explained by SVAS alone. In the present case, calcification of the endocardium progressed rapidly, and biochemical tests excluded calcium deposition, hyperparathyroidism, vitamin D deficiency, renal failure, allergy, and autoantibodies as the cause. The infant's myocardial enzymes were elevated. The TORCH complex examination showed that cytomegalovirus (CMV), rubella virus, and shingles virus immunoglobin G antibodies were positive. The infant had not been vaccinated, indicating that she was previously infected. Dystrophic cardiac calcification is related to CMV infection, and some scholars have found that CMV-infected mice have localized cardiac calcification, mainly in the right ventricle (13). Other animal experiments (14) have shown that mice with ELN deletion develop cardiac phenotypes attributed to aortic stenosis, including compromised fractional shortening, cardiac enlargement, necrosis, and calcification in the cardiac chambers. Based on the above literature, although there were no other related clinical symptoms in the present case, myocardial damage caused by CMV infection combined with SVAS seemed to be the most likely cause of the rapid and progressive LV endocardial calcification in the present case, although the possibility of genetic susceptibility caused by delection of new heterozygosity cannot be ruled out.
4 ConclusionWe report a case of progressive arterial stenosis accompanied by endocardial calcification in an infant with Williams syndrome (WS). To our knowledge, endocardial calcification in WS has not been documented before. After ruling out other potential pathogenic causes, we believe that myocardial injury due to Cytomegalovirus (CMV) infection compounded by aortic stenosis is the probable cause. Nonetheless, we cannot overlook the possibility that a genetic predisposition due to a newly identified heterozygous deletion may have played a role.
Data availability statementThe raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statementThe studies involving humans were approved by the ethics committee of West China Second University Hospital of Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributionsJZ: Writing – original draft. DL: Writing – review & editing. JC: Writing – review & editing, Funding acquisition.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
This research was supported by the Fundamental Research Funds for the Central Universities, the National Key R &D Program of China (grant numbers: 2017YFC0211705, 2017YFC0113905), the Key R & D Program of Science and Technology Department of Sichuan Province (grant numbers: 2019YFS0403, 2019YFS0037), and the Popularization and Application Project of the Sichuan Health and Family Planning Commission (grant numbers: 17PJ415).
AcknowledgmentsWe thank Emily Woodhouse, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Stanley TL, Leong A, Pober BR. Growth, body composition, and endocrine issues in Williams syndrome. Curr Opin Endocrinol Diabetes Obes. (2021) 28(1):64–74. doi: 10.1097/MED.0000000000000588
PubMed Abstract | Crossref Full Text | Google Scholar
3. Ramírez-Velazco A, Aguayo-Orozco TA, Figuera L, Rivera H, Jave-Suárez L, Aguilar-Lemarroy A, et al. Williams-Beuren syndrome in Mexican patients confirmed by FISH and assessed by aCGH. J Genet. (2019) 98(2):34. doi: 10.1155/2015/903175
Crossref Full Text | Google Scholar
4. Collins RT, Kaplan P, Somes GW, Rome JJ. Cardiovascular abnormalities, interventions, and long-term outcomes in infantile Williams syndrome. J Pediatr. (2010) 156(2):253–8.e1.8. doi: 10.1016/j.jpeds.2009.08.042
PubMed Abstract | Crossref Full Text | Google Scholar
5. Szmyd B, Karuga F, Gach A, Moszura T, Kopala M, Respondek-Liberska M. Complex cardiovascular defects in a male infant with Williams syndrome juxtaposed with the results of a preliminary survey illustrating other patients’ outcomes. Kardiol Pol. (2021) 79(2):188–91. doi: 10.33963/KP.15740
PubMed Abstract | Crossref Full Text | Google Scholar
6. Ahrens-Nicklas RC, Reichert SL, Zackai EH, Kaplan PB. Atypical Williams syndrome in an infant with complete atrioventricular canal defect. Am J Med Genet A. (2015) 167A(12):3108–12. doi: 10.1002/ajmg.a.37288
PubMed Abstract | Crossref Full Text | Google Scholar
7. Nance JW Jr, Crane GM, Halushka MK, Fishman EK, Zimmerman SL. Myocardial calcifications: pathophysiology, etiologies, differential diagnoses, and imaging findings. J Cardiovasc Comput Tomogr. (2015) 9(1):58–67. doi: 10.1016/j.jcct.2014.10.004
PubMed Abstract | Crossref Full Text | Google Scholar
9. Abu-Sultaneh S, Gondim MJ, Alexy RD, Mastropietro CW. Sudden cardiac death associated with cardiac catheterization in Williams syndrome: a case report and review of literature. Cardiol Young. (2019) 29(4):457–61. doi: 10.1017/S1047951119000295
PubMed Abstract | Crossref Full Text | Google Scholar
10. Katt TE, Spicer RL, Yetman AT, Ibrahimiye AN, Hammel JM, Robinson JA. Williams syndrome and neonatal cardiac surgery for congenital single ventricle. JACC Case Rep. (2020) 2(11):1716–9. doi: 10.1016/j.jaccas.2020.05.098
PubMed Abstract | Crossref Full Text | Google Scholar
11. Krajcovic J, Janik M, Adamicova K, Straka L, Stuller F, Novomesky F. Giant cell myocarditis and endomyocardial calcification in a 2.5-month-old infant triggered by excessive maternal alcohol abuse: case study of an unusual association. Pediatr Cardiol. (2013) 34(8):2073–6. doi: 10.1007/s00246-013-0637-0
PubMed Abstract | Crossref Full Text | Google Scholar
12. Yuan Y, Zhou R. An infant with suspected missed diagnosis of Williams syndrome failed weaning off CPB after surgical correction of pulmonary stenosis: a case report and literature review. Perfusion. (2023) 38(1):203–7. doi: 10.1177/02676591211046876
PubMed Abstract | Crossref Full Text | Google Scholar
13. Ritter JT, Tang-Feldman YJ, Lochhead GR, Estrada M, Lochhead S, Yu C, et al. In vivo characterization of cytokine profiles and viral load during murine cytomegalovirus-induced acute myocarditis. Cardiovasc Pathol. (2010) 19(2):83–93. doi: 10.1016/j.carpath.2008.12.001
PubMed Abstract | Crossref Full Text | Google Scholar
14. Lin CJ, Staiculescu MC, Hawes JZ, Cocciolone AJ, Hunkins BM, Roth RA, et al. Heterogeneous cellular contributions to elastic laminae formation in arterial wall development. Circ Res. (2019) 125(11):1006–18. doi: 10.1161/CIRCRESAHA.119.315348
留言 (0)