
記住我

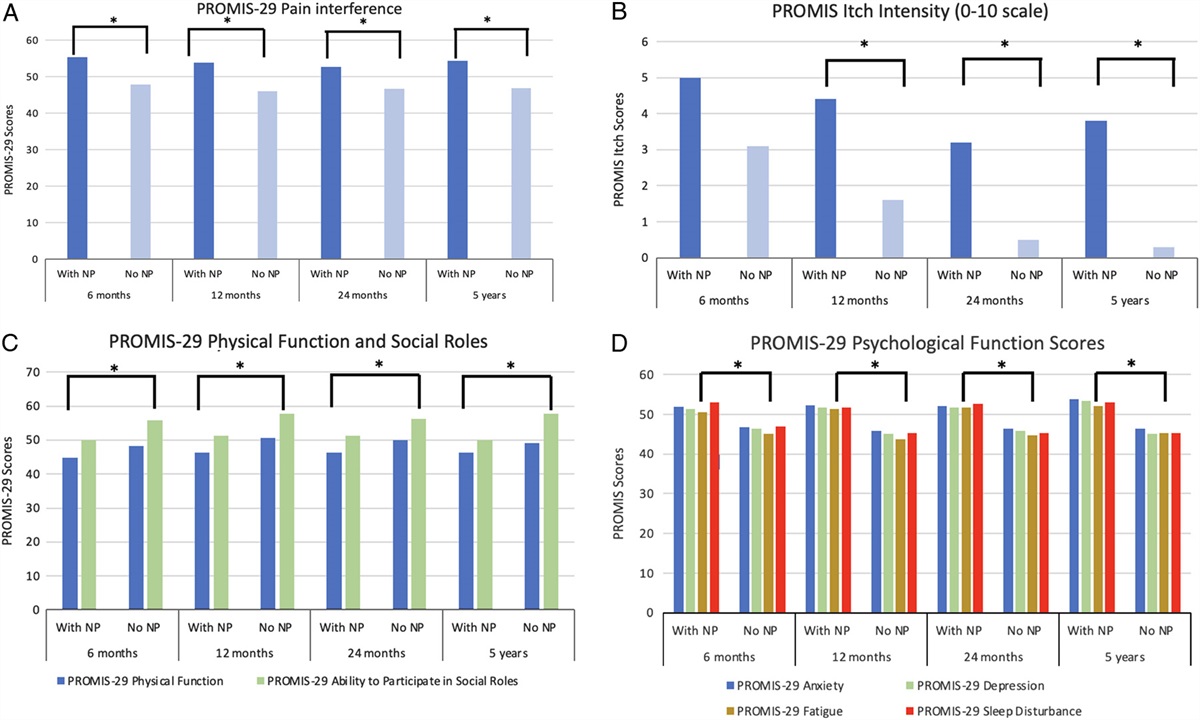

Neutrophils comprise 50% to 75% of leukocytes in the peripheral blood.1–3 Neutrophils are an essential part of the innate immune system and are the first line of defense against invasive microbial pathogens.1–8 When the body is infected by a pathogen, neutrophils marginate, roll via endothelial (E)/platelet (P)/leukocyte (L)-selectin interactions with neutrophil P-selectin glycoprotein ligand-1 (PSGL-1) and tightly adhere to the vascular endothelium via neutrophil β2-integrin family binding with such endothelial ligands as intercellular adhesion molecule, glycosaminoglycans, or fibrin. Neutrophils can then transmigrate from the peripheral bloodstream to infection sites via pseudopodia formation, paracellular diapedesis, and chemotaxis. This is dependent on the reorganization of the neutrophil actin cytoskeleton and intracellular microtubule formation.1,4
Rolling and P-selectin interactions are not required for neutrophil extravasation in pulmonary microvasculature or hepatic sinusoids.1,4 Hepatic sinusoids contain fenestrated or discontinuous endothelium, with areas of direct exposure of extracellular matrix (ECM), which facilitates adhesion and extravasation. Constitutive neutrophil extravasation is a feature of lymphoid tissues containing high endothelial venules, including lymph nodes, lymphoid follicles, and omental milky spots. Extravasated, activated, or adherent neutrophils can engage and destroy pathogens using an arsenal of weapons, including phagocytosis, degranulation and release of cytotoxic enzymes, and oxidative burst. Oxidative burst involves activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase with the generation of reactive oxygen species (ROS).1–8 In 2004, a novel mechanism by which neutrophils attack pathogens was described: the formation of neutrophil extracellular traps (NETs).3
Apart from their canonical role of protecting against disseminated infection, NETs have subsequently been shown to have numerous pathological effects. These include prolonged wound healing, microcrystal diseases, systemic inflammatory response, venous and arterial thromboembolism, impaired immunosurveillance, and promotion of cancer metastases.1–8 Postoperative inflammation, cytokine release, platelet and neutrophil activation, and NET formation may continue for a considerable time after major surgery and potentially be modified by perioperative interventions.3,7 This narrative review aimed to examine the current pathophysiology, prevention, and potential treatment of NETosis in surgical conditions.
NEUTROPHIL EXTRACELLULAR TRAPSNETs are extracellular mesh-like structures formed by extruded decondensed neutrophil chromatin, comprising double-stranded DNA and histones H1, H2A, H2B, H3, and H4.1,2 This DNA mesh is decorated with bactericidal proteins, including neutrophil elastase (NE), myeloperoxidase (MPO), cathepsin G, proteinase 3 (PR3), azurocidin, lactoferrin, gelatinase, lysozyme C, calprotectin, neutrophil defensins, and cathelicidins.1–8 Other proteins and cells may also bind onto this sticky scaffold, including chemotaxins, growth factors, and fibrin.2 NETs exert antimicrobial activity by forming a physical barrier to entrap microorganisms, immobilizing them, and preventing their wider dissemination throughout the body.1,3,9 Furthermore, the NET structure facilitates a high local concentration of antimicrobial peptides and proteins at the site of infection,1–8 including positively charged lysine-rich histones (H1, H2A, and H2B) and arginine-rich histones (H3 and H4). These histones bind negatively charged gram-negative bacterial cell wall lipopolysaccharides (LPSs) and teichoic acid and lipoteichoic acid in gram-positive bacteria. They also stimulate the release of proinflammatory cytokines and the recruitment of immune cells.1–8
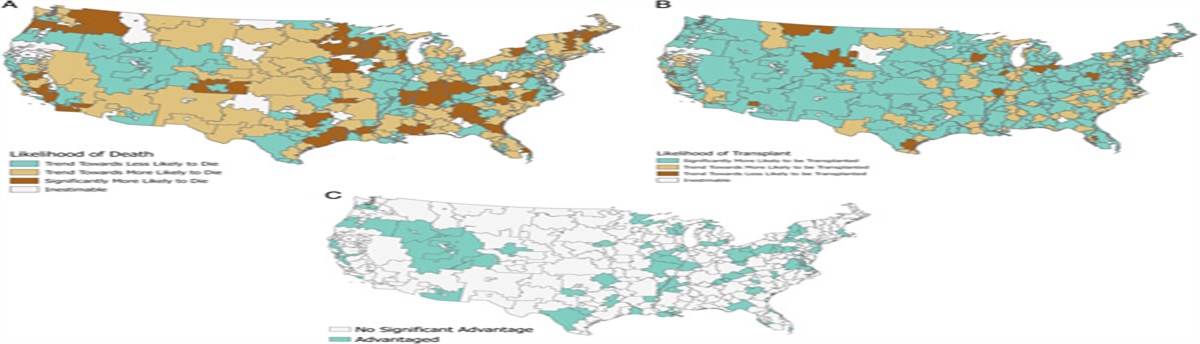
MECHANISM OF NETOSISNETs can arise via 2 pathways.2 The predominant pathway is a neutrophil cell death program, termed lytic or suicidal NETosis.2 This pathway is mainly driven by the generation of ROS from a NADPH oxidase (NOX)-dependent pathway, or mitochondrial-derived ROS (mROS), a NOX-independent pathway.5 The alternative pathway does not involve neutrophil death and is called vital NETosis2,5 (Fig. 1).
 FIGURE 1: Activating stimuli and molecules involved in the 2 types of NETosis: Lytic NETosis (left) consists of a mechanism that effectively kills neutrophils, releasing the filamentous lattice composed of decondensed chromatin, histones, and lytic enzymes into the extracellular space. Known activators of lytic/suicidal NETosis are phorbol 12-myristate 13-acetate (PMA) and antibodies that bind to the Fc receptor (Fcγ-R), which leads to calcium-dependent activation of NADPH oxidase and the release of reactive oxygen species (ROS). ROS activate the PAD4 enzyme and its translocation from granules to the nucleus. Neutrophil Elastase (NE) and myeloperoxidase (MPO), and the combined action of PAD4, NE, and MPO results in citrullination of histones, particularly Histone 3 (cit-H3), and subsequent chromatin decondensation. The nuclear membrane of neutrophils breaks, and chromatin mixed with enzymes and histones is released first into the cytoplasm and then into the extracellular space, following the rupture of the cell membrane, forming Neutrophil Extracellular Traps (NETs). In vital NETosis (right), neutrophils remain intact, releasing the reticulum via a system of vesicles; the latter mechanism appears to be independent of NADPH oxidase activation. Microbial infections, especially from S. aureus, recognized by Toll-Like Receptor-2 (TLR2) or complement receptor and lipopolysaccharide (LPS)-activated platelets that bind TLR-4 are among the major activators of this second pathway. Reproduced from Ronchetti et al with permission.5LYTIC/SUICIDAL NETOSIS
NOX-Dependent NET Formation
FIGURE 1: Activating stimuli and molecules involved in the 2 types of NETosis: Lytic NETosis (left) consists of a mechanism that effectively kills neutrophils, releasing the filamentous lattice composed of decondensed chromatin, histones, and lytic enzymes into the extracellular space. Known activators of lytic/suicidal NETosis are phorbol 12-myristate 13-acetate (PMA) and antibodies that bind to the Fc receptor (Fcγ-R), which leads to calcium-dependent activation of NADPH oxidase and the release of reactive oxygen species (ROS). ROS activate the PAD4 enzyme and its translocation from granules to the nucleus. Neutrophil Elastase (NE) and myeloperoxidase (MPO), and the combined action of PAD4, NE, and MPO results in citrullination of histones, particularly Histone 3 (cit-H3), and subsequent chromatin decondensation. The nuclear membrane of neutrophils breaks, and chromatin mixed with enzymes and histones is released first into the cytoplasm and then into the extracellular space, following the rupture of the cell membrane, forming Neutrophil Extracellular Traps (NETs). In vital NETosis (right), neutrophils remain intact, releasing the reticulum via a system of vesicles; the latter mechanism appears to be independent of NADPH oxidase activation. Microbial infections, especially from S. aureus, recognized by Toll-Like Receptor-2 (TLR2) or complement receptor and lipopolysaccharide (LPS)-activated platelets that bind TLR-4 are among the major activators of this second pathway. Reproduced from Ronchetti et al with permission.5LYTIC/SUICIDAL NETOSIS
NOX-Dependent NET Formation
Of the various pathways that form NETs, NOX-dependent pathways are the best described.6 NOX2 is an enzyme complex located in neutrophil plasma and phagosome membranes.1,6 Complex signaling pathways in neutrophils can result in the activation and assembly of NOX2, with the Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK)/protein kinase C (PKC) pathway being a crucial upstream regulator of suicidal NETosis.1 Stimulation of neutrophil surface membrane receptors activates second messenger signaling via phospholipase C and diacylglycerol (DAG), leading to the release of calcium ions from endoplasmic reticulum stores. The plant toxin phorbol 12-myristate 13-acetate (PMA) enters the neutrophil independently of receptors and simulates the effect of second messenger DAG signaling. The rise in cytoplasmic ionic calcium activates PKC. PKC phosphorylates NOX2 subunits (p47-phox) while suppressing apoptosis by activating anti-apoptotic B-cell lymphoma 2 family proteins, including myeloid leukemia 1 protein.1,2 Another NOX-dependent NETosis pathway involves the activation of c-Jun N-terminal kinase by bacterial LPSs.1 When NOX2 is phosphorylated and activated, neutrophils are driven toward suicidal NETosis rather than apoptosis.1–6
Phosphorylated NOX2 reduces molecular oxygen by transferring electrons from NADPH across membranes.1,6 This generates the superoxide radical (O2•−), which is rapidly converted into hydrogen peroxide.1,6 Alternatively, ROS such as superoxides may be formed by electron leakage onto molecular oxygen from the electron transport chain during mitochondrial respiration powered by glycolysis-generated pyruvate.2 NOX-derived ROS may be required for NETosis in contexts when mROS is insufficient (fungal infection, PMA stimulus).2 ROS link upstream regulatory pathways and the cellular machinery driving NET formation.2
ROS, in turn, have multiple downstream effects.2 ROS-mediated glutathionylation of actin and tubulin is necessary for cytoskeletal changes during NETosis. ROS mediate the release of serine proteases (NE, cathepsin G, PR3, neutrophil serine protease 4, and azurocidin) and the heme-containing MPO into the cytosol from azurophilic neutrophil granules, where they are normally sequestered in resting neutrophils.1–8 Cytosolic NE binds to F-actin and degrades the intracellular actin cytoskeleton, which effectively blocks phagocytosis and commits the neutrophil to lytic NETosis.2 NE then translocates to the nucleus via passive diffusion, a process which is inhibited by dectin-1–mediated phagocytosis of small pathogens.2 NE partially cleaves nuclear histones, acting on lysine-rich and arginine-rich C-terminal histone tails.2 NE, together with the binding of MPO and nuclear protein DEK, promotes chromatin depolymerization and swelling.1,2,6 Neutrophil nuclear envelope and cell membrane permeability are compromised by the assembly of gasdermin D pores.2 Gasdermin D pores exist in a positive feedback loop with NE: NE promotes the activation of gasdermin D, which in turn enhances NE release from neutrophil granules.2 Permeability of the cell membrane increases, and it eventually ruptures, with decondensed DNA released into the extracellular milieu, forming NETs with antimicrobial action.5 Disruption of the cell membrane results in neutrophil death (Figs. 1, 2).
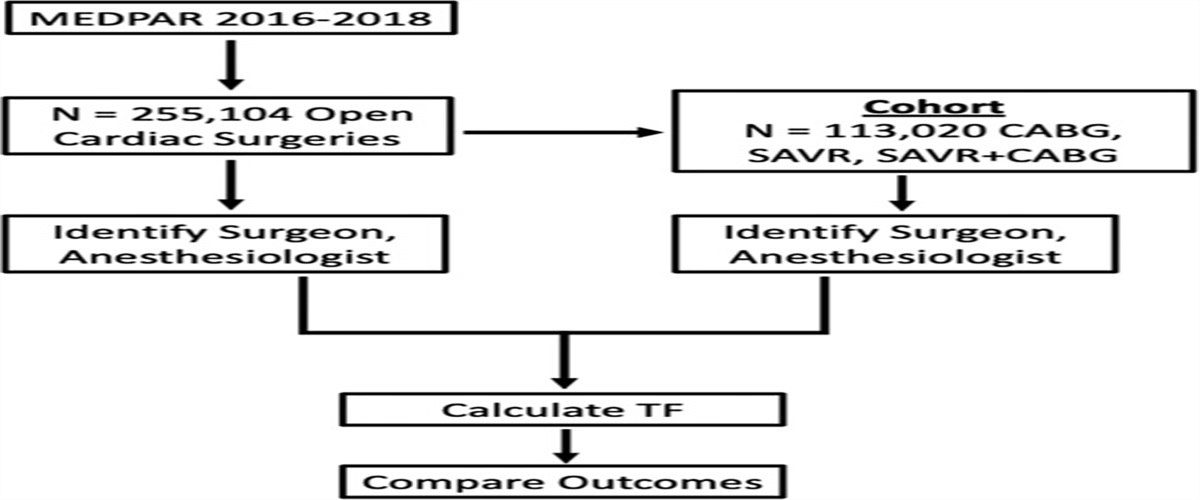
 FIGURE 2: Pathways and mechanisms regulating lytic NETosis. NETosis is triggered by microbial and endogenous stimuli via several activating molecules such as RAGE, PSGL1, TLR, fragment crystallizable-gamma immunoglobulin receptors (Fcγ-R) or sialic acid-binding immunoglobulin-type lectins (Siglec), among others. Activation of MAP kinase signaling induces ROS generation by the NADPH oxidase 2 (Nox2). Alternative ROS can be generated by mitochondria. ROS plays a central role in NETosis triggering NE release from the azurosome complex, a process aided by gasdermin D (GSDMD), which is activated by caspase-11 upon exposure to intracellular cytosolic bacteria. NE degrades F-actin and translocates to the nucleus where it will partially cleave histones promoting chromatin decondensation. Chromatin decondensation is also enhanced by the binding of cationic proteins like MPO or DEK and by PAD4-mediated histone citrullination. Phosphorylation of the lamin network drives its disassembly and the breakdowns of the nuclear envelope. High levels of ROS promote DNA damage triggering DNA repair via ATM and BRCA-1. NETosis also depends on cell cycle CDK4/6 and the duplication of centrosomes and autophagy. Inhibitory receptors such as Siglec-5,9 or SIRL1 block NEtosis. Phagocytic receptors like Dectin-1 inhibit NETosis in response to small microorganisms by sequestering NE to phagosomes. ATG7 indicates autophagy-related protein 7; ATM, ataxia-telangiectasia mutated; AZU, azurophilic granule; BRCA, BReast CAncer gene; CDK4/6, cyclin-dependent kinase 4/6; CG, cathepsin G; CR3, complement receptor 3; GSDMD, gasdermin D; IRAK, IL-1 receptor-associated kinase; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; RIPK1/3, receptor-interacting serine/threonine-protein kinase 1/3; Siglec-5,9, SIRL1, signal inhibitory receptor on leukocytes 1. Reproduced from Hidalgo et al with permission.2NOX-Independent NET Formation
FIGURE 2: Pathways and mechanisms regulating lytic NETosis. NETosis is triggered by microbial and endogenous stimuli via several activating molecules such as RAGE, PSGL1, TLR, fragment crystallizable-gamma immunoglobulin receptors (Fcγ-R) or sialic acid-binding immunoglobulin-type lectins (Siglec), among others. Activation of MAP kinase signaling induces ROS generation by the NADPH oxidase 2 (Nox2). Alternative ROS can be generated by mitochondria. ROS plays a central role in NETosis triggering NE release from the azurosome complex, a process aided by gasdermin D (GSDMD), which is activated by caspase-11 upon exposure to intracellular cytosolic bacteria. NE degrades F-actin and translocates to the nucleus where it will partially cleave histones promoting chromatin decondensation. Chromatin decondensation is also enhanced by the binding of cationic proteins like MPO or DEK and by PAD4-mediated histone citrullination. Phosphorylation of the lamin network drives its disassembly and the breakdowns of the nuclear envelope. High levels of ROS promote DNA damage triggering DNA repair via ATM and BRCA-1. NETosis also depends on cell cycle CDK4/6 and the duplication of centrosomes and autophagy. Inhibitory receptors such as Siglec-5,9 or SIRL1 block NEtosis. Phagocytic receptors like Dectin-1 inhibit NETosis in response to small microorganisms by sequestering NE to phagosomes. ATG7 indicates autophagy-related protein 7; ATM, ataxia-telangiectasia mutated; AZU, azurophilic granule; BRCA, BReast CAncer gene; CDK4/6, cyclin-dependent kinase 4/6; CG, cathepsin G; CR3, complement receptor 3; GSDMD, gasdermin D; IRAK, IL-1 receptor-associated kinase; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; RIPK1/3, receptor-interacting serine/threonine-protein kinase 1/3; Siglec-5,9, SIRL1, signal inhibitory receptor on leukocytes 1. Reproduced from Hidalgo et al with permission.2NOX-Independent NET Formation
Suicidal NETosis can occur without the initial activation of NOX2. NOX-independent pathways are driven by extracellular Ca2+ influx, which can be stimulated by fungal ionophores (nigericin, ionomycin) and granulocyte-macrophage colony-stimulating factor (GM-CSF).8 In turn, calcium ions activate the small-conductance calcium-activated potassium 3 channel (SK3). SK3 mediates the production of mROS, which induces NET formation via peptidyl arginine deiminase 4 (PAD4).1 Stimulated by calcium ion influx and mROS, PAD4 translocates from the cytosol to the nucleus, where it deiminates arginine residues to citrulline in H1, H3, and H4 histones, which reduces their positive charge.1 Thus, histone citrullination weakens the electrostatic bond between histones and the negatively charged DNA backbone, causing chromatin decondensation and swelling, nuclear envelope rupture, and NETosis.1 The role of PAD4 in NOX-dependent NETosis remains contentious.6
VITAL NETOSISA small subset of neutrophils may adopt an alternative pathway termed vital NETosis, in which vesicles containing chromatin and antimicrobial proteins are released into the extracellular space and assembled into NETs without neutrophil cell death.1–8 This is a very rapid (5–60 min), tightly controlled process that is stimulated by bacterial infection.1 However, the exact mechanism underlying this process remains unclear. It appears that bacteria such as Staphylococcus aureus, bacterial LPS, and LPS-activated platelets stimulate vital NETosis by toll-like receptor 2 (TLR2) and TLR4 activation via a NOX-independent pathway.1,6 It has been proposed that vesicles are filled with nuclear DNA and exported through the intact cell membrane by exocytosis.2 Alternatively, vital NETosis can occur when only mitochondrial DNA from neutrophils is released, which requires the formation of oxidized mitochondrial DNA and is NOX- dependent.1,6,9 Vital NETosis allows neutrophils to maintain normal cellular functions as anuclear cytoplasts after NET release, including crawling, chemotaxis, phagocytosis, and oxidative burst activity.9 It is unclear how neutrophils survive and continue this antimicrobial activity after their enucleation.1 (Fig. 1).
NET BREAKDOWNOnce formed, NETs are removed by macrophages.1,6 Host plasma Deoxyribonuclease 1 (DNase1) degrades NETs, and complement component 1q (C1q) opsonizes the debris that is formed.1,6 Subsequently, NETs are internalized by macrophages via a cytochalasin D-dependent endocytic process. NETs are further broken down in lysosomes.
TRIGGERS FOR NETOSISNET formation requires the activation of neutrophils either by bacterial toxins, hypoxia, or stimulation of plasma membrane receptors. Such neutrophil surface receptors include the following:
G-protein–coupled receptors (GPCR) that are chemoattractant/chemokine/complement receptors: platelet-activating factor receptor, complement component 5a receptor, CXC Motif Chemokine Receptor 1 and -2 (CXCR1/2), Chemokine (C-C motif) receptor 1 and -2 (CCR1/2); Fragment crystallizable-gamma receptors (Fcγ-R) for IgG opsonized antigens or immune complexes; Integrins (Lymphocyte function-associated antigen 1 (LFA-1) αLβ2 integrin, macrophage-1 antigen (Mac-1) αMβ2 integrin, very late activation antigen α4β1 integrin) and selectin ligands (L-selectin, PSGL-1); Cytokine receptors: interleukin 1 (IL-1) receptor, GM-CSFR, interferon alpha receptor, tumor necrosis factor alpha (TNF-α) receptor; Pattern recognition receptors in innate immunity that recognize pathogen-associated molecular patterns and host damage-associated molecular patterns (DAMPs): TLR, receptor for advanced glycation end-products (RAGE), formyl peptide receptors, transmembrane C-type lectin receptors, nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3), cytoplasmic nucleotide-binding and oligomerization domain–like (NOD-like) receptors, intracellular retinoic acid-inducible gene I-like receptors (RIG-I-like receptors).1–8 (Fig. 2).There is a diverse range of stimuli for NETosis, which can be both pathologic and physiological, including cytoplasmic calcium ion influx, ROS, signaling cascades (interleukin-1 receptor-associated kinase (IRAK), c-Jun N-terminal kinase, phosphoinositide 3-kinase/protein kinase B (AKT)/mammalian target of rapamycin, and Raf-MEK-ERK-PKC), foreign or microbial pathogens, exosomes, oncogenes (mutant KRAS), proinflammatory cytokines (IL-1β, IL-8, TNF-α, and GM-CSF) and chemokines (IL-8, CXC ligand (CXCL)), microcrystals, or activated platelet-endothelial interactions.1–8 The full range of factors that stimulate NET formation is incomplete, and many new triggers have been discovered, including those involved in sterile inflammation3 (Fig. 2).
PATHOLOGICAL FACTORS Microbial PathogensPathogenic bacteria, viruses, fungi, and protozoa are known to stimulate NET formation, as well as microbial components (bacterial flagellin/LPS, Candida albicans hyphae) and bacterial formyl peptides.1,2 These are recognized by pathogen-associated molecular patterns receptors, including formyl peptide receptor, C-type lectin receptor, TLR-2/4, and NOD-like receptors on neutrophils (Figs. 1, 2). Many microorganisms have developed mechanisms to overcome NETs,3 such as proteolytic enzymes capable of degrading NETs, including extracellular nucleases produced by S. aureus and Streptococcus species.3 Pathogens can also elude NETosis by producing extracellular capsules, such as Pseudomonas aeruginosa and Mycobacterium tuberculosis, or downregulating host inflammatory responses and neutrophil ROS generation (Group A Streptococcus).3
Autoimmune DiseasesActivation of estrogen membrane receptors on neutrophils by 17-β-estradiol, or Fcγ-receptors via immune complexes between IgG and opsonized antigens, have been found to promote NETosis. This is relevant to autoimmune conditions such as systemic lupus erythematosus (SLE) and rheumatoid arthritis and their sexual dimorphism.2 When neutrophils are primed with type I interferon, immune complexes drive NET formation.2 Furthermore, NETs activate the complement system in SLE through the production of C1q, which in turn decreases NET clearance. Auto-antibodies to cell-free (cf) double-stranded DNA, citrullinated proteins, or MPO/PR3 generated by NETosis are respectively implicated in SLE, rheumatoid arthritis, and anti-neutrophil cytoplasmic autoantibody vasculitis.2
CytokinesNETs are involved in postoperative inflammation, systemic inflammatory response syndrome and end organ injury.2,3 Proinflammatory cytokines such as IL-1β, IL-6, IL-8, IL-17A, TNF-α, and C-reactive protein (CRP) induce NETs via neutrophil membrane cytokine receptors and NOX-dependent pathways.2 Interleukins (IL-8) increase cytoplasmic levels of ROS, mediated by nuclear factor kappa B (NF-κB) activation via the CXCR2-phosphoinositide 3-kinase-Protein Kinase B pathway and inducible nitric oxide synthase and cyclo-oxygenase 2 (COX2) induction by NF-κB.2,3 Prostaglandin E2 can be a context-dependent anti-inflammatory mediator and inhibits NET production by increasing intracellular cyclic adenosine monophosphate.2 Activated protein C (aPC) also inhibits NETosis via interactions with the endothelial cell protein C receptor and cleaves histones, which form the backbone of NETs.
Neutrophil Adhesion, Integrins, and PlateletsThe dispensability of neutrophil adhesion and integrin activation in NET formation has been debated. Integrin activation and neutrophil adhesion appear dispensable for exogenous peroxide, calcium influx, or PMA-induced NETosis. However, it is required for viral, LPS, S. aureus, and activated platelet-induced NETosis.2 Direct cell-cell interactions may also regulate NETosis, as attachment to vascular endothelium ligands via neutrophil β2-integrin receptors facilitates NETosis.2 NET release can be achieved by TLR2 and TLR4 activation of platelets, which promotes the binding of platelet P-selectin to neutrophil PSGL-1. Activated platelets can thus relay signals such as the presence of LPSs to neutrophils, triggering NET release. Activated platelets also release soluble ligands, including CCL5, platelet factor 4 (PF4/CXCL4), and high mobility group box-1 (HMGB1). These can bind to neutrophil GPCRs and induce NETosis. Neutrophil activation involves the binding of neutrophil αMβ2 integrin (Mac-1) to its counter-receptor glycoprotein 1bα on platelets and is both proinflammatory and prothrombotic.2
Crystal DiseasesEndogenous inducers of sterile lytic NETosis include microcrystals such as those present in gout [monosodium urate (MSU)], pseudogout (calcium pyrophosphate dihydrate), cholelithiasis (cholesterol), atheromatous plaques (cholesterol), and pancreatitis (calcium carbonate).2 Crystal diseases share a common pathway involving inadequate neutrophil phagocytosis, DAMPs recognition, and activation of NET formation. This is further discussed below.
HOST PHYSIOLOGICAL FACTORSNET release is influenced by host factors, including patient age, gut microbiome, and circadian rhythm.2 Not all neutrophils are equally predisposed to release NETs.2 NET release varies across species, tissues, and within the same organism in different physiological states.2 In the same physiological state, activation of human neutrophils results in NET formation in only 60% of the cells.2 The reason for these differences in response is not fully understood. It is hypothesized that as neutrophils exit the peripheral bloodstream by crossing the basement membrane and circulating through tissues, they become primed, which is permissive for NETosis.2
Host Microenvironment and HomeostasisPhysiological conditions, including pH, O2 content, hyperglycemia, and osmolarity, can modulate NET formation.2 Moderately alkaline conditions cause increased NET formation after stimulation with PMA, uric acid microcrystals, or bacterial LPSs.11 An increase in extracellular pH (>7.4) raises the intracellular concentration of calcium ions, enhancing the production of mROS, PAD4 citrullination of histone 3 (cit-H3), histone cleavage, and NOX-independent suicidal NETosis.10,11 In contrast, a decrease in pH inhibits NETosis, possibly by inhibiting NE/MPO activity or glycolysis. The local oxygen concentration has been shown to influence NETosis.6 Stabilization of the hypoxia-inducible factor 1 (HIF-1α) protein, the major transcription factor that regulates adaptation to hypoxic conditions, promotes NETosis, whereas knockout of the HIF1A gene suppresses NETosis.6 Hyperglycemia or hyperosmotic stress stimulates NETosis by increasing neutrophil intracellular calcium ions.12
NETOSIS and PATHOLOGYAlong with its canonical antimicrobial functions, NETosis can exacerbate inflammation, resulting in tissue damage and contributing to numerous diseases.2 Thus, a fine balance between NET formation and clearance is required to maintain homeostasis.2 If this balance is disturbed by an abnormal increase in NET production or a decrease in NET clearance, pathological NETosis can result.2
CRYSTAL DISEASES CholelithiasisCholelithiasis is a common disease, with an estimated prevalence of 25% in European Caucasians and 60% to 70% in Native Americans over the age of 50.13,14 Furthermore, cholelithiasis and its complications, including acute cholecystitis, common bile duct stones, cholangitis, and gallstone pancreatitis, result in substantial morbidity and mortality.13,14
The mechanisms that lead to cholelithiasis development have been studied but remain incompletely elucidated.14 Bile is excreted by hepatic biliary canalicular cells and is composed of water, lipids, bilirubin conjugates, proteins, and electrolytes.14 The lipid component includes bile salts, phospholipids (>96% of which are mixed phosphatidylcholines), and cholesterol.14 Gallbladder luminal epithelial cells concentrate and acidify the bile to increase the solubility of cholesterol and calcium salts.13 When bile becomes super-saturated and its solubilization capacity is overwhelmed, calcium salts and cholesterol crystals precipitate, forming gallstones.13,14 This is frequently the result of excessive cholesterol secretion and inadequate bile salt/phospholipid excretion from the liver. This is influenced by estrogen, obesity, hyperlipidemia, ethnicity, Western diet, female sex, multiparity, increasing age, and decreased hepatic cholesterol 7α-hydroxylase activity.13,14
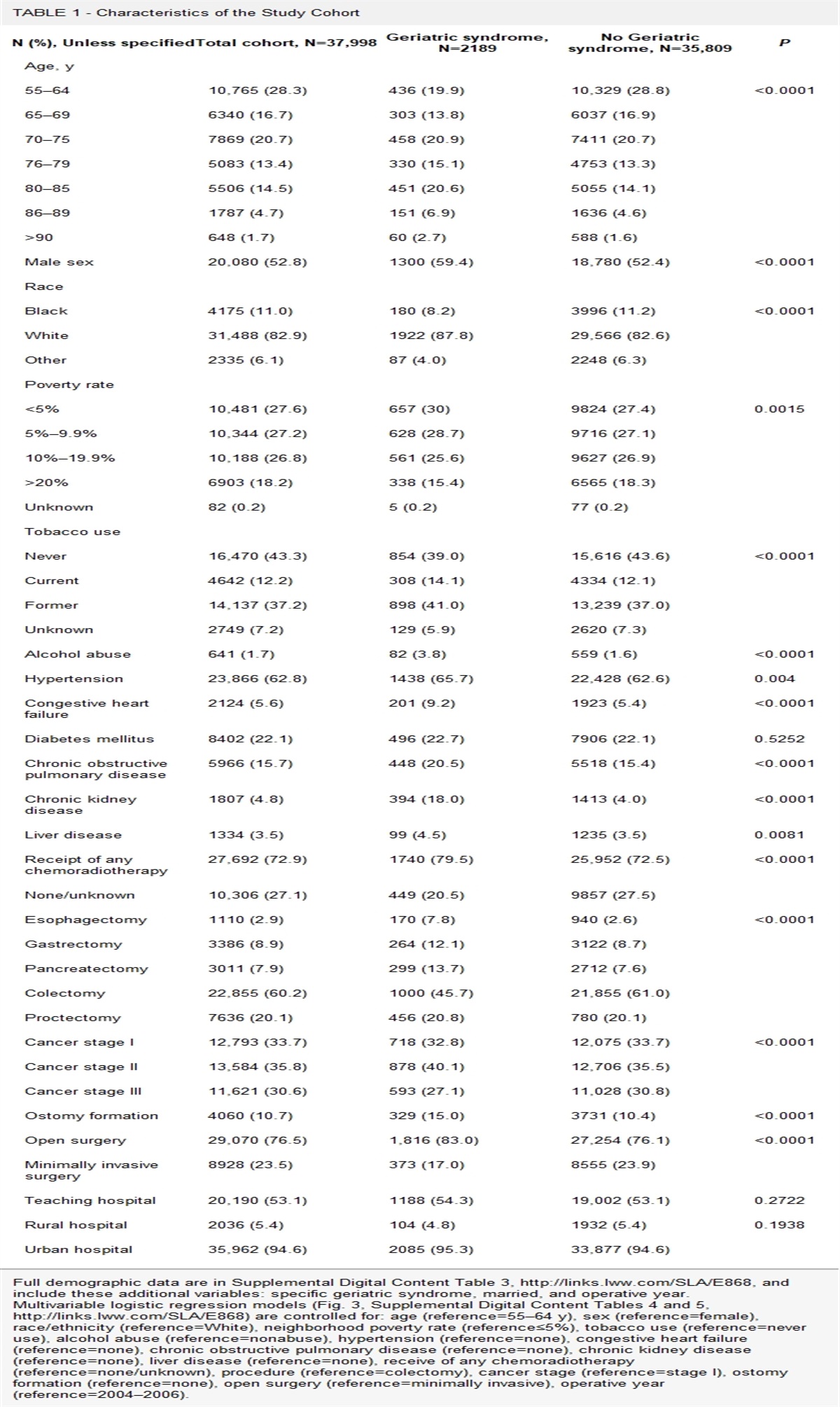
Muñoz et al13 demonstrated that NETs act as aggregation factors contributing to gallstone formation.13 After coming into contact with calcium salts or cholesterol crystals, aggregated NETs form, becoming the “glue” that binds the components of the developing gallstone together. The key mechanism of this process is neutrophil macropinocytosis of calcium and cholesterol crystals and ROS formation.13 Failure of neutrophils or macrophages to successfully complete phagocytosis of large cholesterol crystals stimulates NETosis.13 The successive lamination of cholesterol and NE then leads to gallstone development. The formation of aggregated NETs in developing gallstones was observed using fluorescence microscopy and revealed deposits of extracellular DNA (ecDNA), large ecDNA aggregates, and NE activity.13 In larger gallstones, microscopy showed ecDNA patches on the surface of the gallstones.13 The highest NE activity in the majority (88%) of gallstones analyzed was found at the gallstone surface (Fig. 3).13
 FIGURE 3: Composite macrophotograph of human gallstones, previously immersed in an aqueous propidium iodide solution, under oblique white light (left) and 488 nm light (right) illumination, respectively; extracellular DNA deposits appear in red color. Shown is 1 out of 3 independent experiments. Scale bar, 1 cm. Reproduced from Munoz et al with permission.13
FIGURE 3: Composite macrophotograph of human gallstones, previously immersed in an aqueous propidium iodide solution, under oblique white light (left) and 488 nm light (right) illumination, respectively; extracellular DNA deposits appear in red color. Shown is 1 out of 3 independent experiments. Scale bar, 1 cm. Reproduced from Munoz et al with permission.13The precise mechanism by which neutrophils enter the biliary system and initiate this process has not been fully described. However, in both animal models and humans, the formation of cholesterol gallstones is preceded by inflammation of the gallbladder wall, including edema and the presence of inflammatory cells.12,13 Furthermore, bacteria have been implicated in the pathogenesis of both cholesterol and pigmented stones. A possible mechanism contributing to gallstone formation is the NLRP3 inflammasome.15 The NLRP3 inflammasome is triggered by cholesterol crystals in atherosclerosis and MSU crystals in gout, as well as other endogenous crystals (calcium oxalate, calcium pyrophosphate dihydrate, and cystine) and exogenous crystals (asbestos, silica, aluminum adjuvant, and titanium dioxide).2,13,15
Cholesterol Crystals and NETosisDuewell et al16 demonstrated that cholesterol crystals taken up by macrophages are incompletely degraded in phagosomes and transferred to lysosomes, where they induce the rupture of the phagolysosomal membrane. This results in the release of the lysosomal cysteine protease cathepsin B, which acts as a signal to prime and activate the NLRP3 inflammasome.15 Undegraded cholesterol crystals released upon phagolysosomal membrane rupture may also act as DAMPs sensed by pattern recognition receptors, resulting in activation of the innate immune system.15 Cholesterol crystals thus induce inflammation via “frustrated phagocytosis” and NLRP3 inflammasome activation, with IL-1β and IL-18 release.15 In turn, this inflammation attracts neutrophils to the site of cholesterol crystals, resulting in potent stimulation of neutrophils by interleukin-1 receptor-associated kinase and Receptor Interacting Protein Kinases (RIPK1–RIPK3)-mixed lineage kinase domain-like protein signaling and NET formation.2
Pancreatitis and Pancreatic Ductal StonesIn addition to their role in gallstone formation, NETs contribute to acute pancreatitis and pancreatic ductal calcium carbonate stones in chronic pancreatitis.17 Pancreatic secretions contain alkaline bicarbonate ions and calcium carbonate crystals, which can strongly stimulate NET formation via PAD4 activation.2,13 Pancreatic ductal occlusion caused by aggregated NETs can drive acute pancreatic inflammation.2,13 In a murine model of severe acute pancreatitis, NETs induced trypsin activation, inflammation, and tissue injury in vivo.17 Cell-free histones derived from NETs disrupt the pancreatic acinar cell plasma membrane, resulting in acinar cell death. Interactions between activated platelets and NETs within the pancreas also cause thrombus formation and injury to the pancreatic microvasculature, leading to pancreatic necrosis. Systemic manifestations of severe acute pancreatitis associated with NETosis include acute lung injury, vascular damage, venous thrombosis, increased vascular permeability, and acute kidney injury. NETs also contribute to increased disease severity in patients with pancreatic ductal stones.17
GoutAcute exacerbations of recurrent gout are common (17.2%–44.3%) after major surgery, related to fasting, systemic acidosis, blood transfusion, volume depletion, tissue hypoxia, or presurgical hyperuricemia. The risk of perioperative gout is especially increased in males, patients with obesity or diabetes mellitus, previous gout history, high-protein purine-rich diets, after abdominal, orthopedic or bariatric surgery, or perioperative cessation of colchicine gout prophylaxis.18 In acute gout, needle-like MSU microcrystals deposited in tissues and joints are incompletely phagocytosed by monocytes and neutrophils, causing activation of the NLRP3 inflammasome, production of IL-1β, and further neutrophil recruitment.13,18 Neutrophils are stimulated by MSU crystals and macrophage-derived IL-1β to form aggregated NETs, which are known to be present in gouty tophi.13 While NETs contribute to tophi formation by densely packing MSU crystals with ecDNA, aggregated NETs can trap and cleave proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokines (CCL-2) in acute gout.13,17 Thus, NETs can contribute to acute inflammation and tophi formation but also play a role in the resolution of inflammation in gout.13,17
TreatmentsMuñoz et al13 demonstrated that interfering with NET formation using neutrophil cytosolic factor-1/p47-phagocyte oxidase (Ncf1**/p47-phox)-deficient or PAD4-deficient mice models reduced both the prevalence and size of gallstones formed in the setting of a lithogenic diet. Pharmacological treatment of wild-type mice with a PAD4 inhibitor (GSK484) or the selective β1-adrenergic receptor antagonist metoprolol, which is known to stun neutrophils, also had this effect.13 Metoprolol and GSK484, in combination, completely abrogated gallstone progression.13 Colchicine inhibited microtubule-dependent neutrophil macropinocytosis of cholesterol crystals and prevented aggregated NET formation. Thus, NET inhibition was successful in preventing gallstone formation and growth.13 Inhibition of NETosis by colchicine treatment is also effective in other microcrystal diseases, including gout, pseudogout, and atherosclerosis. Colchicine binds to tubulin and inhibits microtubule assembly, which is required for neutrophil chemotaxis, adhesion, transmigration and recruitment, GPCR signaling, Ca2+ influx, NOX2 superoxide production, intracellular transport, nuclear chromatin swelling, organelle trafficking, and NETosis.13,18 Acute perioperative gout can pose difficulties with arthritis severity, pain management, and patient immobilization. Colchicine is a useful intervention, due to its efficacy and the relative contraindications of perioperative systemic corticosteroid or nonsteroidal anti-inflammatory drug administration.18 PAD4 inhibition or DNase1 therapy was shown to decrease the severity of pancreatic and lung injury in murine models of severe acute pancreatitis.17
POSTOPERATIVE WOUND HEALINGWound healing is an important concern of surgeons. Altered wound healing and complications can cause significant morbidity and mortality during the postoperative period.3 Wound healing is a coordinated physiological process that restores the skin barrier function. The phases of wound healing include hemostasis, inflammation, proliferation, and remodeling.19 Neutrophils play a crucial role in this process; during the inflammatory phase, they are recruited to the wound site and provide defense against microbial pathogens. However, numerous factors may affect normal wound healing. These include local factors, such as ischemia, infection, presence of a foreign body, and edema; and systemic factors, including diabetes mellitus, sepsis, medications, and obesity.19–21 Although neutrophils provide crucial protection against infection, if inflammation is dysregulated by excessive NETs, delayed wound healing results.2
The deleterious effects of NETs are mediated through multiple mechanisms. NET-derived NE degrades essential wound proteins, including proteoglycans, collagen, and fibronectin, disrupting cell-cell interactions.19–21 The presence of excessive NETs in wounds impairs angiogenesis and healing by promotion of endothelial-mesenchymal transition through the Merlin/Yes-associated protein (YAP)/HIPPO/mothers against decapentaplegic homolog 2 (SMAD2) pathway.19 NE present within NETs cleaves platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), also hindering angiogenesis.3 NETs may increase angiogenesis when in contact with platelets and endothelial cells; however, in chronic wounds, NETs can have an inhibitory effect.19 Furthermore, endothelial cell function is dysregulated by NET-derived mixed metalloproteinase 9 (MMP-9). Histones exert a local cytotoxic effect by integrating into the phospholipid bilayer of cell membranes, altering membrane permeability, which can result in calcium ion influx and subsequent cell death. Keratinocyte migration and proliferation are necessary for the restoration of the epithelial layer. However, high wound levels of NETs inhibit keratinocyte migration and delay wound healing.21 NETs can induce fibroblast activation and differentiation into myofibroblasts, thereby promoting wound fibrosis.19
Diabetes, Wound Healing, and NETosisThe impairment of wound healing by NETosis is common in patients with diabetes mellitus.2 Diabetes mellitus primes neutrophils to form NETs.2,12 A diabetic mouse model demonstrated higher levels of wound cit-H3, a biomarker of NET formation, and slower rates of wound healing.21 The mechanisms of increased NETosis in diabetes include hyperglycemia, glucotoxicity, increased cytokine/CRP priming of neutrophils, AGE formation via polyol and hexosamine pathways, neutrophil AGE-RAGE/NF-κB signaling and superoxide formation, de novo DAG synthesis, and PKC activation.12 Furthermore, patients with diabetes are predisposed to NETosis due to elevated basal neutrophil calcium levels, which stimulates PAD4 activation. A preliminary study in surgical patients undergoing total joint arthroplasty showed obese patients with insulin-resistance had higher PAD4 expression at the surgical site than insulin-sensitive control patients.20
TreatmentsCurrently, anti-NET treatments are being developed to promote wound healing.21 The most studied treatment is DNase1, which degrades NETs. In a murine study, the recombinant human DNase1 (Dornase alfa) reduced the wound area and enhanced re-epithelialization by 75%, with accelerated wound healing.21 By inhibiting NETosis, PAD4 knockdown achieved faster wound healing at 2 weeks compared with wild-type mice (80% vs 25% healed).21 A matrix of Thr-Asp-F-amidine tripeptide, a PAD4 inhibitor, with the standard wound healing materials alginate and gelatin was applied to wounds.21 Wounds treated in this manner showed accelerated re-epithelialization and improved healing compared to controls or wounds treated with alginate and gelatin alone.21 Intravenous treatment with Cl-amidine, which inhibits PAD4, also decreases wound area. In addition to its anti-hyperglycemic effect, metformin has been shown to inhibit the PKC-NOX pathway, ROS generation, and NET formation.21,22 Therefore, metformin could have a positive effect on the promotion of wound healing and is an area that requires further investigation.
CHRONIC WOUNDSIn addition to postoperative wounds, NETosis also affects chronic wounds and secondary intention healing, including bacterial biofilm formation. Extracellular DNA is a major component of bacterial biofilms.23–25 Such ecDNA can originate from neutrophils via NETosis or neutrophil necrosis or from bacteria.24In vitro, ecDNA acts as a structural component of bacterial biofilm extracellular polymeric substance23,24 as well as a diffusion barrier that increases the resistance of bacteria to cationic antibiotics (aminoglycosides and colistin).25 A recent in vivo study of Pseudomonas aeruginosa biofilms showed that host-derived ecDNA acted as a protective shell overlying the biofilm but was not found inside the biofilm.24 It was thought that because the neutrophil-derived ecDNA on P. aeruginosa biofilms in vivo contained H3 but not cit-H3, it was derived from neutrophil necrosis rather than NETosis.24 Not all NETs contain cit-H3, and NETosis can occur in the absence of histone citrullination. Thus, further studies of other bacterial in vivo biofilms are required.2,8,23
TreatmentsRecombinant DNase1 has been investigated as a potential treatment for chronic wounds and biofilms, including pleural empyema, surgical wounds, and diabetic foot ulcers.2,3 The use of maggot therapy with anti-NET/DNase properties should also be explored. Topical maggot therapy facilitates debridement of necrotic, gangrenous, or infected tissues, has antimicrobial effects, and stimulates wound healing.26,27 The debridement of both acute and chronic wounds is an important aspect of wound management.26,27 Debridement takes different forms, including surgical, biological (eg., maggot therapy), mechanical (eg, wet-to-dry dressing changes, negative pressure wound therapy), enzymatic (eg, streptokinase in hydrogel), and autolytic (eg, manuka honey).26,27 Debridement facilitates wound healing by the direct removal of all devitalized tissue, reduction of bacterial burden and biofilm, and stimulation of granulation tissue. However, biofilm recurrence after surgical debridement can occur within 3 days.27
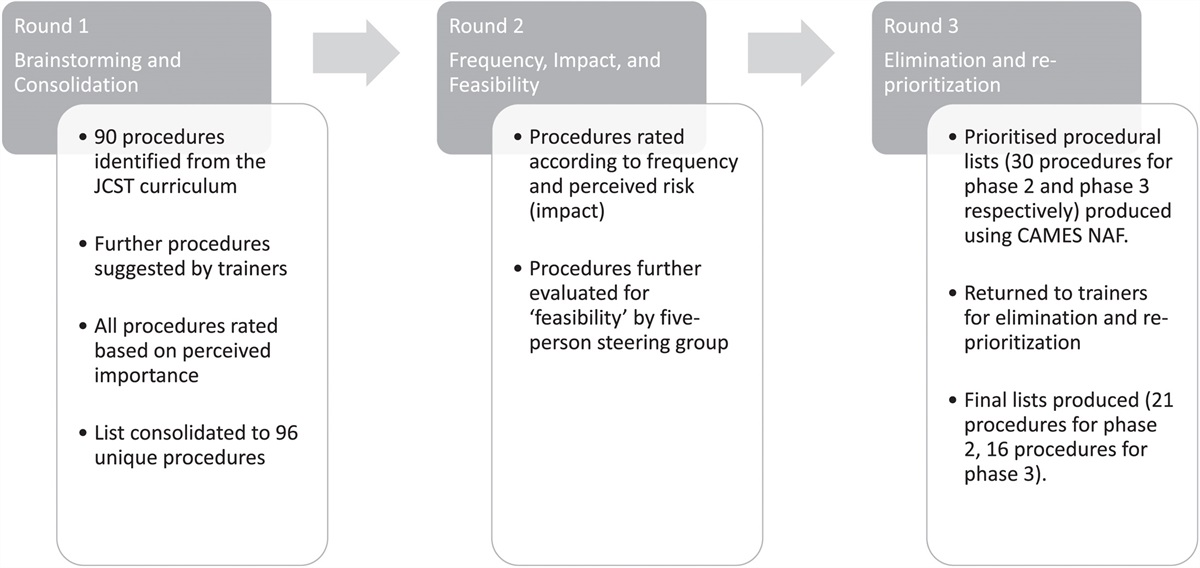
Maggot debridement therapy (MDT) for infected or chronic wounds has been used since antiquity.26,27 The larvae of the green bottle blowfly Lucilia sericata are used in sterile medicinal MDT, which involves multiple mechanisms.26,27 Firstly, maggots scrape the wound base as the larvae move across the surface of the wound, loosening and feeding on non-viable tissue and bacteria.27 Maggots secrete digestive enzymes that liquefy necrotic tissue and slough in the wound, including a wide range of MMPs and serine proteases.27 Maggots not only digest bacteria in a wound but secrete and excrete bactericidal compounds with activity against aerobic and anaerobic gram-positive bacteria and some gram-negative bacteria.27 Maggots also release DNases, which can effectively degrade both microbial and human DNA within necrotic debris, potentially explaining their ability to remove biofilms and prevent biofilm reformation.26,27 This disruption of the bacterial biofilm is synergistic with systemic antibacterial therapy, as neutrophil-derived ecDNA is used to form a capsule around the biofilm, preventing antibiotic penetration.26 MDT has been shown to be superior to conventional treatments for chronic wounds in comparative studies (including 6 RCTs), with faster healing, longer antibiotic-free periods, and lower amputation risk (Fig. 4).27
 FIGURE 4: Maggot therapy with free-range larvae. Diabetic foot ulcers during and after MDT. Photographs by Parizad et al.27THROMBOSIS
FIGURE 4: Maggot therapy with free-range larvae. Diabetic foot ulcers during and after MDT. Photographs by Parizad et al.27THROMBOSIS
NETs interact with platelets, complement (C5), and tissue factor (TF) and activate both the intrinsic and extrinsic coagulation cascades to promote venous thromboembolism (VTE) and arterial thrombosis and occlusion.5,9,22,28–36 (Fig. 5) Risk factors for VTE include malignancy, major surgery, hypoxia, obesity, pregnancy, infection, i
留言 (0)