
記住我




Psychiatric and cognitive disorders is a global issue for modern society. According to data from the World Health Organization for the year 2022, such disorders occur in 1 out of 8 people (World Health Organization, 2022). There are multiple theories laying the grounds for the development of cognitive and psychiatric disorders; however, most of them pinpoint an imbalance between excitatory and inhibitory structures in the brain as the primary reason (Walsh et al., 2008). Such an imbalance can be caused by dysfunction of the nervous system (deficiency in an enzymatic system responsible for neurotransmitter production, disturbance of neurotransmitter secretion and reuptake, defects in the structure and function of neurotransmitter receptors, channelopathies) as well as organic damage or developmental abnormalities in the brain. Furthermore, as supported by numerous genetic and twin studies, heredity is considered an underlying cause for a variety of psychiatric and cognitive disorders such as schizophrenia, endogenous depression, affective disorders and autism (Kallmann, 1946; Gardner and Stephens, 1949; Detera-Wadleigh et al., 1987; Egeland et al., 1987; Gassó et al., 2015).
The human brain, serving as the biological substrate for consciousness and cognitive functions, is extraordinarily intricate. It typically consists of around 100 billion neurons, alongside a multitude of neuroglial cells that provide support to these neurons. Each neuron establishes multiple connections with up to 10,000 other neurons, transmitting signals through approximately 1,000 trillion synapses (Stiles and Jernigan, 2010).
The proper functioning of the brain is ensured by correct morphological and structural development of different brain parts as well as precise synaptic connections of individual neurons. The development of such a complex structure relies on the sophisticated orchestration of multiple cellular and molecular mechanisms, since their disruption or imbalance can have devastating consequences and result in a vast array of neurodevelopmental and cognitive disorders. A prime example of this principle in action is schizophrenia. In several studies, the authors reported a perturbation of spatial neuron positioning, reduced neuron density and density of neural connection in the hippocampus, prefrontal cortex, and anterior cingulate gyrus (Grafton et al., 1992; Tamminga et al., 1992). In autism spectrum disorders deviations in brain development have also been reported, typically manifesting as uneven and disproportional brain growth, enlarged white matter relative to gray matter, and a delayed gray matter formation (Torun et al., 2015; Lukito et al., 2020). Of note, autosomal recessive primary microcephaly is associated with abnormalities in the survival and proliferation of embryonic neural progenitors (Stiles and Jernigan, 2010). Aberrations in embryonic corticogenesis is related to neurodevelopmental disorders such as lissencephaly, megalencephaly, schizophrenia, Rett Syndrome, intellectual disorders, Down syndrome, and meningeal heterotopias (Costa et al., 2007; Meyerink et al., 2020). All of those mentioned above ascertain the core importance of correct embryonic brain structure development for its normal functioning and mental health in the adult individuum (Walsh et al., 2008; Stiles and Jernigan, 2010; Gassó et al., 2015; Ishii et al., 2016; Lukito et al., 2020). Comparing to other theories of the innate nature of mental disorders, the neurodevelopmental theory of the origin of psychiatric and cognitive disorders is significantly underestimated. Since comprehensive reviews on the subject are still lacking, here we aim to fill in the gap and congregate the existing data on the potential mechanisms of neurodevelopmental abnormalities as well as on the key neurotrophic factors and guidance molecules related to these pathological conditions. Detailed research (including the stage of prenatal development) may allow for potential mental disorder prediction as well as provide a solid ground for new strategies, such as gene therapy, to guide future treatment-prevention approaches (Sasi et al., 2017).
2 Key stages of neurodevelopment, guidance molecules, and secreted factorsHuman brain development starts in the third week of gestation and continues until late adolescence. It involves neurogenesis, neuronal migration, establishment of neuronal connection, axonal growth and myelination. Disruption of any of these courses is considered to be responsible for a wide spectrum of pathologies (Johnson et al., 2009; Corroenne et al., 2022). Neurodevelopment is affected by gene expression as well as by environmental conditions encompassing the presence and variety of information stimuli (Miguel et al., 2019).
2.1 Early stages of human brain developmentTo shed light on the pathogenesis of psychiatric and cognitive disorders, we will briefly consider the underlying neurodevelopmental mechanisms in the brain with a focus on the individual molecules.
Embryonic nervous system development starts with the differentiation of epiblast cells into neural progenitor cells (neuroectodermal cells) as a result of complex molecular signaling that involves multiple gene expression. Sonic Hedgehog (SHH) and antagonists of Bone Morphogenetic Protein 4 (BMP4): chordin, noggin, and follistatin, which are produced by notochord cells, are among the most important signaling molecules (Greene and Copp, 2009; Stiles and Jernigan, 2010; Sears et al., 2022). By the end of gastrulation, the neural progenitor cells are located along the rostral-caudal midline of the upper part of the three-layered embryo forming a structure referred to as a neural plate. The neuroectoderm ridges formed along the sides of the neural plate (E21) rise folding inward and then fuse, starting at the neural plate center and continuing in both, the rostral and caudal parts, forming a hollow neural tube. By the 26th–27th day, the neural tube closes along the entire length. This stage of embryonic development, from the neural plate formation until the neural tube is complete, is known as neurulation (Greene and Copp, 2009; Stiles and Jernigan, 2010).
Neuroepithelial cell proliferation within the walls of the rostral end of the neural tube and the internal pressure of cerebrospinal fluid lead to the formation of three primary brain vesicles: the precursor of the forebrain (prosencephalon—the anterior vesicle), midbrain (mesencephalon—the middle vesicle), and hindbrain (rhombencephalon—the posterior vesicle) (Fame et al., 2020). The rostral-lateral parts of the forebrain grow faster than the others, leading to the formation of two outgrowths (diverticula) in the rostral part—the so-called telencephalon, which further develop into the cerebral hemispheres. The caudal part of the forebrain develops into the diencephalon, which gives rise to the intermediate brain (epithalamus, metathalamus, thalamus, and hypothalamus). The mesencephalon gives rise to the midbrain, and the rhombencephalon divides into two segments: the metencephalon (which gives rise to the pons and cerebellum) and the myelencephalon (which gives rise to the medulla oblongata) (Greene and Copp, 2009; Stiles and Jernigan, 2010; Fame et al., 2020).
Proliferation of the neuroblasts lining the neural tube and their radial outward migration from the central part ultimately results in the architectural complexity and multilayered structure of the neural tube. Different regions of the neural tube exhibit peculiarities reflected in the formation of specific brain and spinal cord segments (Delhaye-Bouchaud, 2001; Barry et al., 2013). Particularly, in the region of the brain vesicles the process results in the formation of the cortex, basal ganglia, and white matter as discussed below in more detail (Delhaye-Bouchaud, 2001; Barry et al., 2013).
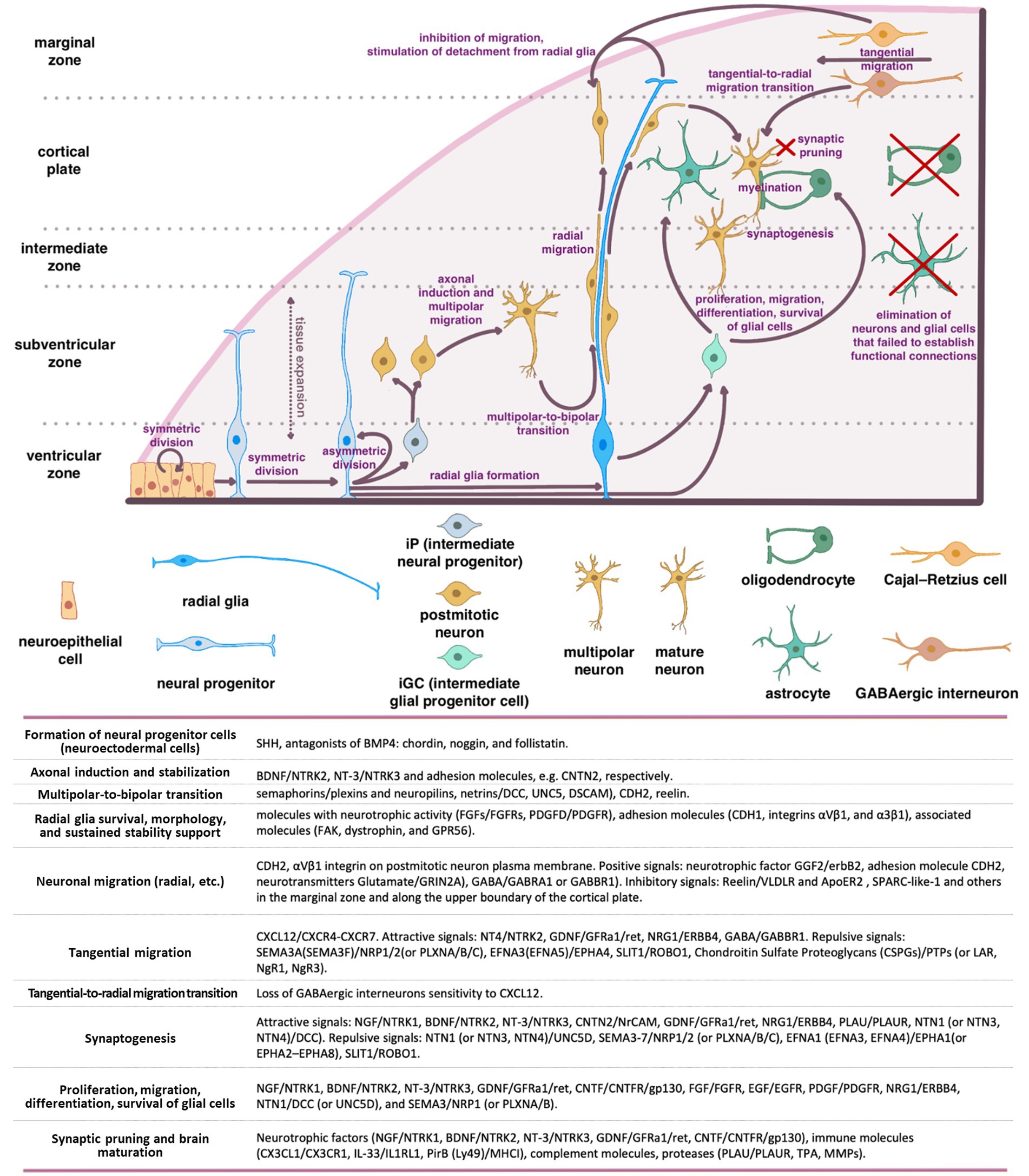
2.2 Migration of neurons in the developing brainNeuronal positioning is crucial for the formation of distinct brain regions such as the cerebral cortex, hippocampus, and cerebellum. In humans, from the end of gastrulation and until E42, the population of neural progenitor cells, located in the ventricular zone (VZ) of neural tube, undergo mitotic divisions, producing identical daughter cells (“symmetric division”) (Figure 1). However, starting from E42, the type of cell division in neural progenitors progressively shifts toward an asymmetric one, resulting in one neural progenitor cell and one neuron. Most of the postmitotic neurons leave the VZ and migrate into the developing cortex. However, a certain population of neurons maintain their position and will later develop into the basal ganglia. At the same time, the neural progenitors remain in the proliferation zone and proceed with the asymmetric divisions (Wodarz and Huttner, 2003; Stiles and Jernigan, 2010).
Figure 1. Schematic representation of the main stages of brain morphogenesis with the indication of key regulatory molecules.
A series of coordinated migrations take place in the developing cortex when neurons migrate from the site of origin to their final destination to form complex dendritic and axonal networks there. Three main types of migration have been described: radial (from the center of the neural tube), tangential (along the surface of the developing brain), and multipolar.
The basis for radial migration are so-called radial glia cells, which established a strong mechanical connection in the early stages of corticogenesis, both with the pial (outer) surface of the cortex and with the basal layer in the ventricular zone (proliferation zone). Migrating cortical neurons physically associate with bipolar radial glia and migrate along with their ascent to the top of the cortex early in corticogenesis. As the cortex grows and thickens, glial cells can stretch for millimeters, guiding the migration of neurons even along the migratory paths with inhibitory signals and layers of the previously migrated neurons (Wodarz and Huttner, 2003; Stiles and Jernigan, 2010).
The first migrating neurons form the preplate—primordial plexiform layer above the ventricular zone, which restricts the outward neuron migration and helps organize the layers of the developing cortex. Subsequently, neurons migrate within the preplate to form four or six layers of the mature cortex (depending on its type) (Rice and Curran, 2001; Cooper, 2008). The second wave of migrating neurons splits the preplate into two separate layers marginal zone (MZ) and subplate (SP). Both the MZ and SP disappear at the end of fetal period (Stiles and Jernigan, 2010).
The neurons that form deeper cortical layers (V–VI) migrate earlier, while those forming the superficial layer (II) migrate later resulting in the “inside-out” pattern of development. Experiments on neural progenitor cell transplantation demonstrated that this migration is strongly affected by the microenvironment, which encompasses a complex interplay of cells, molecules, and blood vessels that surround and provide support to them (Lathia et al., 2007; Kazanis et al., 2008; National Cancer Institute, 2024). Although, the regulatory mechanisms remain unclear, the radial glia to a large extent determines the migratory paths of the neurons and the speed of migration (Rice and Curran, 2001; Cooper, 2008). This form of migration is particularly crucial for pyramidal glutamatergic neurons that populate the appropriate layers of the cerebral cortex (Luhmann et al., 2015).
According to the current concepts of embryonic development, radial migration of postmitotic neurons is normally preceded by their multipolar migration in the subventricular and intermediate zones of the developing brain (Tabata and Nakajima, 2003). Migrating multipolar neurons undergo axonal induction that makes them to protrude an axon that is stabilized by intercellular adhesion molecules (e.g., CNTN2).
BDNF/NTRK2 and NT-3/NTRK3 are among the most important and well-known neurotrophic factors that induce the axonal growth in multipolar neurons. The shift in neuron migration from multipolar to radial one is a critical step (termed a multipolar-to-bipolar transition), which is orchestrated by guidance molecules such as semaphorins (receptors—plexins and neuropilins) and netrins (receptors—DCC, UNC5, DSCAM), as well as by CDH2 and Reelin (Cooper, 2014).
In this intricate process, molecules with neurotrophic activity (including FGFs/FGFRs and PDGFD/PDGFR), adhesion molecules (CDH1, integrins αVβ1, and α3β1), and associated molecules such as focal adhesion kinase (FAK), dystrophin, and GPR56, significantly contribute to supporting radial glia survival, morphology, and sustained stability (Penisson et al., 2019). During radial migration (relevant for other types of neuron migration), neurons are exposed to gradients of positive and negative signals, ensuring their precise positioning in the developing cortex. Among the key positive signals are: neurotrophic factor GGF2 (glial growth factor-2, a soluble form of neuregulin1)/erbB2, adhesion molecule CDH2 (N-cadherin) expressed on the surface of radial glial cells, and neurotransmitters Glutamate (signals via GRIN2A), GABA (signals via GABRA1 or GABBR1) (Anton et al., 1997; Luhmann et al., 2015). Experimental findings provide compelling evidence that CDH2 and αVβ1 integrin expression on the plasma membrane of postmitotic neurons are essentially important for their migration (Meyerink et al., 2020; Rashid and Olson, 2023). Soluble proteins, including Reelin, SPARC-like-1, and others distributed within the marginal zone and along the upper boundary of the cortical plate, provide inhibitory signals that restrict the radial neuron migration (Chai and Frotscher, 2016).
Cajal–Retzius cells (CR) that have colonized the superficial layer of the developing cortex (so-called marginal zone) produce Reelin, that triggers the detachment of radial migrating neurons from radial glia, inhibits migrating neurons from exiting beyond the cortex, and instructs them to take up proper positions within the forming cortical plate. To date, two receptors for Reelin on the surface of migrating neurons have been identified: very low-density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2) (Dlugosz and Nimpf, 2018).
SPARC-like-1 operates in a similar manner, inducing the detachment of migrating neurons from the radial glial cell processes at the upper boundary of the cortical plate. The source of SPARC-like-1 is believed to be the radial glial cells themselves, with its expression and secretion being particularly prominent in the apical segment of radial glial cell processes. As of today, a receptor for SPARC-like-1 on the surface of neurons remains undefined (Dlugosz and Nimpf, 2018).
Cajal–Retzius cells (CR) primarily originates from the cortical hem, ventral pallium, and pallial septum. Unlike neurons migrating from the VZ, the primary type of CR migration is tangential, traversing long distances along the surface of the cortex. This type of migration enables CR to be the initial cell population of the superficial marginal zone in the developing cortex. CR orchestrate the subsequent colonization of the cortical plate by postmitotic neurons undergoing radial migration (Paxinos and Marín, 2015). Modern concepts of brain morphogenesis consider CR cells as a transient population within the cerebral cortex. Once they have served their purpose, these cells undergo programmed cell death (Elorriaga et al., 2023).
The tangential migration is also recognized as a predominant mode of migration for GABAergic interneurons (INs). These neurons hold a crucial role in synchronizing the activity of excitatory glutamatergic pyramidal neurons in the cerebral cortex and organizing the transmitted information (Llorca and Deogracias, 2022). Any misplacement or dysfunction of these interneurons may potentially contribute to the development of certain psychiatric disorders.
The rodent cerebral cortex comprises three primary types of interneurons (INs): parvalbumin-positive (PV+), somatostatin-positive (SST+), and 5HTR3a-positive neurons. PV+ INs establish connections with the soma, proximal dendrites, and proximal axon segments of pyramidal neurons. SST+ INs target the distal dendrites of pyramidal neurons, interact with basket-like PV+ INs within cortical layer IV, and form reciprocal connections with layer IV Purkinje cells (Naka et al., 2019). Additionally, 5HTR3a+ INs predominantly populate the surface layers of the neocortex, exerting inhibitory effects on other INs and thereby mediating the disinhibition of pyramidal neurons (Pfeffer et al., 2013; Jiang et al., 2015; Favuzzi et al., 2019).
Cortical INs originate from distinct compartments within the ventral subpallium: the medial ganglionic eminence (MGE) and preoptic area (POA) give rise to PV+ and SST+ INs. In contrast, the caudal ganglionic eminence (CGE) produces 5HTR3a+ INs, while the lateral ganglionic eminence (LGE) serves as the primary source of GABAergic projection neurons for the striatum (Llorca and Deogracias, 2022).
IN precursors initially emerge from neural progenitors located in the ventricular and subventricular zones of the subpallium. Subsequently, they migrate to their designated layers within the developing brain, such as the marginal zone (MZ), cortical plate, or they may remain within the subventricular zone and undergo tangential migration. Notably, PV+ and SST+ INs primarily migrate within the MZ, and their migration trajectory is determined by their origin and cellular identity. During migration through various regions of the developing cortex, INs encounter molecular cues that determine their fate, phenotype, and electrophysiological characteristics—a process often referred to as spatial and temporal patterning (Butt et al., 2007; Jovanovic and Thomson, 2011; Allaway et al., 2021). Once they arrive at their designated cortical regions, they undergo radial migration to integrate into the developing neural circuits (Llorca and Deogracias, 2022).
Tangential migration is defined by a diverse array of signaling pathways distinct from the radial migration. Of note, the CXCL12/CXCR4-CXCR7 signaling axis plays a pivotal role in guiding the tangential migration of GABAergic interneurons (Llorca and Deogracias, 2022). Loss of sensitivity to CXCL12 triggers a transition from tangential to radial migration, resulting in their entry into the developing cortical plate and the establishment of connections with pyramidal neurons (Marín, 2013; Luhmann et al., 2015). In addition to the CXCL12/CXCR4-CXCR7 axis, a wide spectrum of neurotrophic and guidance signals are involved in tangential migration. These signals encompass both attractive cues, such as BDNF/NTRK2, NT4/NTRK2, GDNF/GFRa1/ret, HGF/c-Met, VEGFA/FLT1(FLT4, FLK1, NRP1, NRP2), NRG1(NRG3)/ERBB4, GABA/GABBR1, dopamine/D1, and repulsive signals including SEMA3A (or SEMA3F)/NRP1/2 (or PLXNA/B/C), EFNA3 (or EFNA5)/EPHA4, SLIT1(SLIT2)/ROBO1, FLRT2(FLRT3)/Unc5B (Unc5D), chondroitin sulfate proteoglycans (CSPGs)/PTPs (or LAR, NgR1, NgR3), dopamine/D2 (Marín, 2013; Fleitas et al., 2021; Llorca and Deogracias, 2022). The urokinase plasminogen activator (uPA)/urokinase plasminogen activator receptor (uPAR) system plays a significant role in the migration of interneurons (INs). This system is involved in various processes, including extracellular matrix degradation, cell migration, growth cone guidance, and angiogenesis (Semina et al., 2016, 2017; Toudji et al., 2023).
As INs mature and reach their designated cortical regions, their tangential migration halts, and a switch from tangential to radial migration occurs. Loss of sensitivity to CXCL12 coupled with increased sensitivity to GABA (Bortone and Polleux, 2009), along with activation of the NRG3/ERBB4 signaling axis are considered to be the main mechanisms launching tangential-to-radial switch (Llorca and Deogracias, 2022; Toudji et al., 2023). Additionally, microvessels and radial glia have been described as guiding tracks for the radially migrating INs in some studies (Yokota et al., 2007; Léger et al., 2020). Upon reaching the target cortical layer, INs establish connections with pyramidal neurons and other INs. The crucial stages of interneuron origin and development, as well as the main classes of molecules involved in these processes, are shown in the Supplementary Figure S1.
2.3 Establishing the interneuronal connectionsIn tandem with proliferation and migration of neural progenitor and their derivatives, the connections between the developing brain structures are established. This facilitates the coordination of brain structures activity and integrates the brain into a unified center for information processing, the center for generating motor and mental activity.
Two of the most important conducting pathways in the brain are the dopaminergic thalamocortical (TCT) and glutamatergic corticothalamic (CTT) tracts (Gasiorowska et al., 2021) because, by transmitting sensorimotor information, they connect the neocortex to sensory organs and target organs. In humans, the development of these pathways occurs between the 20th and 26th weeks of gestation (Kostović and Jovanov-Milosević, 2006). Subplate neurons play a pivotal role in shaping these pathways. Initially, they receive afferent fibers from thalamic nuclei (TCT) and act as guides for the growth of CTT fibers. As TCT (thalamus > neurons L4–L5) and CTT (neurons L5–L6 > thalamus) mature and stabilize, subplate neurons reduce their connections and gradually undergo apoptosis. Disturbances in the activity of subplate neurons or their premature demise can lead to abnormalities in the TCT or CTT formation, potentially causing the neurological and psychiatric disorders (Rubenstein et al., 2020).
Disruptions in dopaminergic pathways in the brain, particularly the mesolimbic (MLT) and mesocortical (MCT) tracts, play a fundamental role in the development of various psychiatric disorders, including schizophrenia, attention deficit hyperactivity disorder (ADHD), and addictions. These tracts establish connections between the ventral tegmental area (VTA) and the nuclei of the limbic system (such as the nucleus accumbens (NAcc), amygdala, and olfactory bulb) as well as with neurons in the prefrontal cortex (PFC) (Prakash and Wurst, 2006). NAcc, in turn, is considered a part of the ventral striatum, where the information from the PFC and the limbic system (including the hippocampus and amygdala) is integrated. It is widely believed that the NAcc is responsible for motivating the individuals to achieve specific goals and for seeking new behavioral patterns related to reward. The distinct functions within the NAcc are attributed to its core and shell regions. Experimental studies demonstrated that afferent fibers from the hippocampus and PFC terminate in the NAcc’s shell and core, respectively, exerting reciprocal effects on synaptic plasticity in these parts of the brain (Goto and Grace, 2008). Therefore, an imbalance in afferent signals to the NAcc from the PFC and the limbic system can bring about various psychiatric disorders. For example, a decrease in afferent signals from the PFC can lead to a long-term potentiation in the NAcc’s shell, triggering synaptic remodeling and the activation of reward-related behavioral patterns, potentially contributing to the development of addictions (Goto and Grace, 2008). Furthermore, hyperactivation of the hippocampus, often due to dysfunction or loss of parvalbumin GABAergic interneurons, and the subsequent transmission of excitatory signals from the hippocampus to the NAcc, amygdala, and PFC, are considered to be the key factors of both: positive and negative symptoms emerging in schizophrenia (Goto and Grace, 2008).
While the exact mechanisms underlying the formation and maturation of interneuronal connections remain incompletely understood, it is well-established that the precision and functionality of these connections are regulated by a plethora of neurotrophic and guidance molecules. These guidance molecules comprise both attractive cues, such as NGF/NTRK1, BDNF/NTRK2, NT-3/NTRK3, CNTN2/NrCAM, GDNF/GFRa1/ret, NRG1/ERBB4, PLAU/PLAUR, NTN1 (or NTN3, NTN4)/DCC and repulsive ones including NTN1 (or NTN3, NTN4)/UNC5D, SEMA3-7/NRP1/2 (or PLXNA/B/C), EFNA1 (EFNA3, EFNA4)/EPHA1(or EPHA2–EPHA8), SLIT1/ROBO1 (Niederkofler et al., 2010).
These signaling molecules also determine the development of the corpus callosum, a pivotal structure that provides the functional integration of the cerebral hemispheres. Disruptions in the corpus callosum development can give rise to seizures, as well as intellectual, coordination and psychiatric disorders (Niederkofler et al., 2010).
Radially migrating INs establish GABAergic connections with pyramidal neurons and other INs. The signaling axes of NRG3/ERBB4, BDNF/NTRK2, and NT4/NTRK2 are considered to play a key role in this process, as their disruption or malfunctioning increases excitability and oscillatory activity of pyramidal cells, as well as impaires the synchrony of oscillations between distinct cortical areas. In mutant mice, these functional disorders manifest as heightened locomotor activity, aberrant emotional responses, compromised working memory, altered social behavior, and impaired cognitive functions—symptoms specific to certain mental disorders (Curley and Lewis, 2012; Lewis et al., 2012; Del Pino et al., 2013). Interneurons (INs) that fail to establish functional connections undergo cell death (Llorca and Deogracias, 2022).
2.4 Development of brain glia and myelination of nerve fibersNeurons need protection, support, and trophic supply, while nerve fiber need proper electrical isolation (myelin sheath) for efficient and rapid transmission of signals within and beyond the brain. In the brain, these vital functions are fulfilled by two distinct types of glial cells: oligodendrocytes and astrocytes.
Intermediate glial progenitor cells (iGCs), which are precursors of astrocytes and oligodendrocytes, are generated through the division of neural stem cells or radial glial cells located in the subventricular zone of the forebrain. These iGCs subsequently migrate to various regions including the white matter, cortex, striatum, and hippocampus, where they undergo differentiation into astrocytes and oligodendrocytes (Stiles and Jernigan, 2010). Notably, recent studies revealed the presence of glial progenitors in the marginal zone (MZ) of the developing cortex, potentially contributing to the upper cortical layers (Costa et al., 2007). Upon reaching their respective destinations, these glial progenitors establish connections with neurons and undergo differentiation to oligodendrocytes or astrocytes (Weng et al., 2019). Oligodendrocytes are responsible for myelinating the dendrites and axons, thereby facilitating the action potential conduction and providing neurotrophic support. Astrocytes, in turn, offer trophic support to neurons, participate in establishing the blood-brain barrier (BBB), and contribute to the formation and operation of functional synapses. The proliferation, migration, differentiation, and survival of glial cells are regulated by a set of key molecules, including NGF/NTRK1, BDNF/NTRK2, NT-3/NTRK3, GDNF/GFRa1/ret, CNTF/CNTFR/gp130, FGF/FGFR, EGF/EGFR, PDGF/PDGFR, NRG1/ERBB4, NTN1/DCC (or UNC5D), and SEMA3/NRP1 (or PLXNA/B) (Klämbt, 2009; Pöyhönen et al., 2019). Impairments in proliferation, migration of glial cells, or their dysfunction alter information transmission in the brain and are frequently linked to the onset of cognitive and psychiatric disorders (Zambon et al., 2017; Zhou et al., 2019; Ishikawa et al., 2020).
2.5 Maturation of the nervous system and elimination of non-functional connectionsAn essential stage in brain development is eliminating the neurons and glial cells that failed to establish functional connections with each other and did not integrate into neural networks (Rakic and Zecevic, 2000; Buss and Oppenheim, 2004). Additionally, synaptic pruning of non-functional (or excessive) synaptic connections occurs. This phenomenon arises from the competition of the established cells for limited resources, mostly for neurotrophic factors. Insufficient availability of these factors results in the demise of neurons and glial cells (Huang and Reichardt, 2001). Neurotrophic factors such as NGF/NTRK1, BDNF/NTRK2, NT-3/NTRK3, GDNF/GFRa1/ret, and CNTF/CNTFR/gp130, along with immune molecules like CX3CL1/CX3CR1, IL-33/IL1RL1, PirB (Ly49)/MHCI, and complement molecules, as well as proteases including PLAU/PLAUR, TPA, and MMPs, all play pivotal roles in brain maturation and synaptic pruning (Levi-Montalcini, 1964; Oppenheim et al., 1989; Faust et al., 2021). Microglial cells and astrocytes are currently recognized as the principal agents responsible for the natural process of synaptic pruning (Faust et al., 2021). Physiological death of non-functional neurons and cell populations (such as SP and MZ cells, including CR cells) that regulate migration and wiring predominantly takes place during the prenatal period.
An imbalance in the expression of these neurotrophic factors or disruptions in their signaling pathways results in excessive neuronal or glial cell death or, conversely, survival, which can lead to the onset of cognitive and psychiatric disorders (Walsh et al., 2008; Stiles and Jernigan, 2010; Bathina and Das, 2015; Angoa-Pérez et al., 2017; Zambon et al., 2017; Zhou et al., 2019; Ishikawa et al., 2020).
In the postnatal period, brain development persists with processes such as proliferation and migration of glial precursors, axon myelination, elimination of non-functional glial cells (those not connected to neurons), and synaptic pruning (Hughes, 2021). The list of key molecules governing these processes is detailed above (Levi-Montalcini, 1964; Oppenheim et al., 1989; Faust et al., 2021).
Therefore, the process of shaping the brain structures and their integration into a cohesive entity is intricate and multi-phased. Proliferation, migration, regulated cell death of neural precursors, glial cells, and transient cell populations, as well as the establishment of interneuronal connections, and myelination of nerve fibers are all orchestrated by an ensemble of molecular and cellular signals to ensure the correct development and maturation of the brain. At the same time the functions of individual brain regions are determined already at early stages of development. All together this provides a remarkable level of reliability in formation of functional and appropriately interconnected structures within the central nervous system, however, any dysfunction or malfunction of key coordinators in this process can result in disruptions to nervous system development and function (Bishop et al., 2002; Walsh et al., 2008; Stiles and Jernigan, 2010; Gassó et al., 2015).
A comprehensive study of these molecular families and the intricacies of their intracellular signaling is beyond the scope of the present review, given that the previously published literature is widely available (Levi-Montalcini, 1964; Oppenheim et al., 1989; Klämbt, 2009; Niederkofler et al., 2010; Pöyhönen et al., 2019; Faust et al., 2021). Our primary focus here is on data reflecting the dysfunction of key coordinator molecules and regulators of brain formation (neurotrophic and guidance molecules) in the development of mental and cognitive disorders.
3 Growth factors and guidance molecules implicating the impaired brain development and onset of cognitive and psychiatric disordersThe morphogenetic theory of mental disorders was first proposed by the Scottish psychiatrist Clouston (1891), and recent studies have only expanded the list of proteins and genes, whose dysfunction can lead to inappropriate formation of brain structures, the loss or aberrant inter-neuron connections ultimately manifesting in psychiatric and cognitive disorders.
3.1 Shh/Ptch signaling pathwayThe Shh/Ptch signaling pathway is one of the first involved in the formation of the neuroectoderm, which gives rise to the nervous system. Therefore, loss-of-function mutations of the Shh gene, malfunction of its receptors or associated signaling cascades can lead to severe nervous system malformations that are often incompatible with life. These abnormalities include holoprosencephaly, cyclopia, the reduced number or absence of ventral cell types in the spinal cord, and anomalies of midline structures (Chiang et al., 1996).
Severe developmental defects leading to prenatal embryonic death are also associated with dysfunction of the BMP4 antagonist chordin (Chrd gene), resulting in abnormal development of the embryo in the ventrodorsal direction, ventralization of its tissues, and disruption of neuroectoderm formation (Bachiller et al., 2003; Troilo et al., 2014). As can be seen from the data described above, null mutations in Shh/Ptch or Chrd genes are lethal during the prenatal or early postnatal period. However, in some cases, non-lethal mutations such as VAR_023804 (R6T), rs760920236-C (S15W), rs28936675-T (G31R), and others in the SHH gene (The UniProt Consortium, 2023a), and rs145871696-G (T457S) in the CHRD gene (The UniProt Consortium, 2023b) downregulate the activity of these proteins, disrupt the formation of neuroectoderm, and may lay the basis for the onset of mental and cognitive disorders (holoprosencephaly, schizencephaly, autism, and others) (Nanni et al., 1999; Roessler et al., 2009). The genomic variants discussed in this review are summarized in Supplementary Table S1.
3.2 Neurotrophins and their receptorsNeurotrophins comprise a family of neurotrophic growth factors closely related to nerve growth factor (NGF). Within mammals, this family consists of four proteins: NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5). Neurotrophins are key molecules for sustaining proliferation and survival of neural progenitors and glial cells, as well as for the formation and maturation of inter-neuron connections, have also been shown to be involved in the manifestation of mental disorders (Mitre et al., 2017). For instance, in a number of patients with schizophrenia, a decrease in BDNF protein and mRNA has been detected in the hippocampus, prefrontal cortex, anterior cingulate cortex, and superior temporal gyrus (Hashimoto et al., 2005; Issa et al., 2010; Ray et al., 2011, 2014; Ray et al., 2014; Thompson et al., 2014), as well as the reduction of NTRK2 and NTRK3 (neurotrophin receptors) mRNA in the dorsolateral prefrontal cortex (Weickert et al., 2005). A lack of neurotrophins in embryonic tissues is observed during pregnancy in women with severe mental disorders (Freedman et al., 1992, 1994; Bersani et al., 2000), which reduces the proliferation and survival of neural progenitors during brain morphogenesis and can underlie the onset of mental illness or cognitive dysfunction. Moreover, studies have established a correlation between the incidence of single nucleotide polymorphisms (SNPs) in the genes NGF, BDNF, NTRK1, NTRK2 and the susceptibility to paranoid schizophrenia (Arévalo et al., 2004, 2010; Lin et al., 2013; Kranz et al., 2015).
BDNF (brain-derived neurotrophic factor, BDNF gene), a classical neurotrophin, is recognized to be one of the key molecules affecting brain volume (Freedman et al., 1994). BDNF is expressed by both neurons and glial cells and, as mentioned above, is a molecule with broad neuroprotective activity. It controls the proliferation, survival, migration, and apoptosis of neural progenitors and mature neurons (including sensory and cholinergic neurons in the basal forebrain, mesolimbic dopaminergic neurons, hippocampal neurons, granular neurons of the cerebellum, etc.). BDNF is also involved in neurogenesis, synaptogenesis, and stabilization of the inter-neuron connections in the brain (Segal et al., 1992; Freedman et al., 1994; Sasi et al., 2017; Ping et al., 2022).
Impairments in BDNF expression and maturation may inflict the manifestation of cognitive and mental disorders at different levels: via the dysfunction of the brain dopaminergic system caused by dopaminergic neuron death, or via alterations in the plasticity/stability of inter-neuron networks. At the same time, BDNF gene is not included in the list of loci associated with the onset of schizophrenia (Pardiñas et al., 2018), which is due to the fact that the data on BDNF and predisposition to schizophrenia are inconsistent.
In 2020, Chinese scientists Ping et al. (2022) analyzed the association of the BDNF gene SNPs [rs11030101-A (intr), rs2030324-A (intr), and rs6265-C (V64V)] with predisposition to schizophrenia and found no significant correlation. However, when patients were subdivided using the positive and negative syndrome scale for schizophrenia (PANSS) scale, an association of rs1103010101-A with negative symptoms of schizophrenia and rs6265-C with attention deficit was determined. Analysis of BDNF gene haplotypes revealed that the rs11030101/rs2030324/rs6265 AAC haplotype was more common in patients with schizophrenia than in healthy individuals, and that negative symptoms were more pronounced in rs11030101-A homozygous patients.
One of the most studied BDNF polymorphisms, rs6265-T (V66M), which impairs activity-mediated BDNF secretion (Tsai, 2018), has been shown to be associated with a decrease in bilateral hippocampal volume (independent of patient’s sex and age) compared to the V66V homozygotes from the control group (Tate et al., 2012, 2015). Val/Met heterozygous individuals also showed the reduced lateral convexity of frontal cortex gray matter volumes (Tate et al., 2015). A study by Kranz et al. (2015) identified another missense variant rs769727156-T (G198D) in the BDNF gene in patients with schizophrenia-related psychosis (American population); however, its functional significance in predisposition to psychiatric and cognitive disorders remains to be established.
A number of other BDNF polymorphisms have been correlated with the manifestation of major depressive disorder—rs12273539-T (intr), rs11030103-G (intr), rs6265-T (V66M), rs28722151-G (intr), rs41282918-T (3′UTR), and rs11030101-T (intr) (in a Mexican American population) (Licinio et al., 2009), and Alzheimer’s disease (in a Japanese population)—rs56164415 (C270T) (Kunugi et al., 2001). In a Chinese population, the occurrence of the dinucleotide polymorphism (GT)n in BDNF gene was found to correlate with early manifestation of schizophrenia and sensitivity to chlorpromazine treatment (Xu et al., 2008).
Due to the complexity of the nervous system structure and species-specific peculiarities of intracellular signaling, the functional significance of the BDNF polymorphisms and their putative mechanisms underlying the predisposition to mental disorders are yet to be unveiled (Gören, 2016).
The defects in the overall neurotrophic system (neurotrophic factors and their receptors) functioning may result from the altered signaling downstream the corresponding receptors. Polymorphisms and mutations in these receptor genes, impairing their expression or signaling activity may cause the dysregulated signaling. Overall, these defects may ultimately lead to neurological and psychiatric disorders. For example, in a Polish population the polymorphisms of the BDNF receptor—TrkB (NTRK2 gene): rs10868235-T (intr) and rs1387923-G (3′UTR) was found to be associated with a higher risk of schizophrenia in men; rs1387923-A (3′UTR)—with a lower risk of schizophrenia, and the polymorphism rs1565445-A (intr)—with an increased risk of suicide in schizophrenia (Suchanek-Raif et al., 2020). It is worth noting that many of the described TrkB SNPs are associated with the predisposition to suicidal activity; therefore, it is suggested that NTRK2 polymorphisms should be screened for if there was a history of suicidal ideation. The predisposition to schizophrenia (and other mental disorders) may be determined by a combination of several SNPs within several genes (so-called haplotypes), and their influence may differ between the sexes. For example, the ATAAT haplotype (5 SNP NTRK2) is associated with a reduced risk of schizophrenia in men, and the GTAGCC haplotype [5 SNP NTRK2 and 1 rs6265-C (V64V) SNP of the BDNF gene] is associated with a reduced risk of it in women (Suchanek-Raif et al., 2020). Combinations of mutations in the ligand [BDNF, rs6265-T (V66M)] and receptor [NTRK2, rs1387923-G (3′UTR)] and rs2769605-T [(3′ downstream)] genes can significantly increase the risk of paranoid schizophrenia (in a Chinese population), even though the separate presence of such polymorphisms is not associated with this risk (Tsai, 2004, 2007; Hwang et al., 2006).
NGF (nerve growth factor, NGF gene), the first neurotrophin discovered (Levi-Montalcini, 1987), has been shown to stimulate the survival of sensory, sympathetic, cholinergic and mesencephalic dopaminergic neurons (Mobley et al., 1986; Gibbs, 1994; Hanaoka et al., 1994; Calamandrei and Alleva, 1995; Kang and Schumann, 1996) in the basal forebrain, hippocampus, cerebellum (Thoenen et al., 1987; Whittemore and Seiger, 1987), etc. The highest level of NGF expression is observed in hippocampus, prefrontal cortex (Large et al., 1986; Yan et al., 1997) and pituitary (Goedert et al., 1986; Hefti et al., 1986). Together with BDNF, NGF influences brain development by controlling cell death, fiber growth direction, dendrite morphology, and synapse formation (Thoenen, 1995). Moreover, NGF can induce the onset of mental and cognitive disorders by regulating the dopamine D2-receptor activity (Fiorentini et al., 2002), as well as by impairing oligodendrocyte formation/maturation and myelination (Chan et al., 2004). A number of NGF single-nucleotide genomic variants may be associated with the predisposition to cognitive and psychiatric disorders. Thus, it was shown that SNP variant rs6330-A (A35V) (Licinio et al., 2009; Zakharyan et al., 2014), as well as the haplotype rs12760036-C (intr)/rs4839435-A (intr) (in the Korean population) (Park et al., 2011) increase the risk of schizophrenia onset. The variant rs4565713-G (intr) is associated with susceptibility to schizophrenia manifestations in Russian and Tatar populations (Gareeva et al., 2015), and autism in an American population (Chen et al., 2006; Lu et al., 2013). NGF variants rs2856813-G (intr), rs6678788-T (intr), rs4529705-A (intr), rs6537860-A (intr), rs4332358-T (intr), and rs3811014-G (promoter) are associated with primary affective disorders (PAFDs) in women in the American population (Cui et al., 2011). NGF diplotype GG-TC in rs2856813 (intr) and rs6678788 (intr) has the strongest association with PAFDs in women in the same study (Cui et al., 2011), and the haplotype rs2254527-rs667878788-rs12760036 CCA (all introns) is associated with lower susceptibility to antidepressant therapy in major depressive disorder in a Chinese population (Yeh et al., 2015).
Abnormalities in NGF signaling caused by disruption of the structure of its receptors [TrkA (NTRK1 gene) and p75 (NGFR gene)] or the signaling pathways involved, may also impair brain morphogenesis and lay the groundwork for the onset of mental and cognitive disorders. For example, some of the Ntrk1 genomic variants [rs6336-T (H568Y) and rs4661063-A (intr)] were found to be associated with changes in the microstructure, such as the decreased myelination and impaired fasiculation of nerve fibers of the white matter of the cerebral cortex in rats (Su et al., 2021). The role of NTRK1 in mental disorders is indirectly supported by the data that antipsychotics can reduce the level of TrkA autophosphorylation in the rat hippocampus (Terry et al., 2010), but the mechanism of the pro-psychotic activity of NTRK1 remains to be established. Several lines of evidence show that increased TrkA activation may be associated with the manifestation of psychiatric disorders. For instance, the frequency of rs6336-T (H568Y) variant, which creates an additional potential site of TrkA autophosphorylation, correlates with the predisposition to schizophrenia in American and European populations (van Schijndel et al., 2009, 2011), although rs6336-T (H568Y) variant was identified as a protective factor in a number of experimental cohorts (van Schijndel et al., 2011). On the other hand, reduced TrkA-mediated signaling may also be a risk factor for psychiatric diseases. In particular, the rs556840308-A (G4S) NTRK1 variant (Kranz et al., 2015), that impairs TrkA exposure to the membrane, has been identified in patients with schizophrenia in an American population. Mutations in adaptor proteins mediating signal transduction from Trk receptors may also underlie the pathogenesis of psychiatric diseases. Another example is the rs748531715-C (H1085R) mutation in the KIDINS220 gene, which encodes a scaffold protein and a substrate for Trk receptors, was shown to be associated with severe paranoid hallucinations and schizophrenia manifestation in adulthood (Encinas et al., 2000; Lasky-Su et al., 2008).
Another receptor for neurotrophins is the low-affinity nerve growth factor receptor p75NTR (NGFR gene) (Casaccia-Bonnefil et al., 1999; Bradshaw et al., 2015). This receptor predominantly binds the proforms of neurotrophins (as its affinity for mature neurotrophins is substantially lower) and has the capability to initiate apoptotic cascades in neural cells.
Recent evidence suggests that impaired signaling of neurotrophins through p75NTR may be associated with the predisposition to psychiatric and cognitive disorders. For example, Zhao et al. (2022) showed that NGFR SNPs rs2072446-T (S205L) and rs11466162-A (3′UTR) were associated with the incidence of schizophrenia, and Ngfr knockdown in mice was associated with schizophrenia-like social behavior abnormalities. A study of NGFR SNPs has revealed that rs11466155-T (G265G) and rs2072446-T (S205L) are associated with an increased risk of schizophrenia in an Armenian population. While rs734194-G (3′UTR) appeared to be a protective factor, SNPs rs734194, rs11466155 and rs2072446 had no significant effect on schizophrenia incidence (Park et al., 2011). At the same time, rs2072446-T (S205L) of the NGFR gene was also associated with an increased risk of Alzheimer’s disease and β-amyloid deposition (He et al., 2022). A Spanish population was shown to be amendable to addiction due to rs534561-G (intr) (Fernàndez-Castillo et al., 2013).
Neurotrophin-3 (NT-3, NTF3 gene) has a similar structure as the other neurotrophins (NGF, BDNF) but has a different expression profile in the brain (Ernfors et al., 1990; Maisonpierre et al., 1990a). It is actively expressed in all brain regions of mouse embryos at postnatal day 4 (Thompson et al., 2014), including the hippocampus and neocortex. NT-3 plays an important role in brain maturation (Maisonpierre et al., 1990b; Nimgaonkar et al., 1995), but during adulthood its expression level decreases (Hattori and Nanko, 1995) and in the adult brain it is predominantly expressed in the hippocampus. Along with NGF and BNDF, NT-3 ensures the survival of dopaminergic neurons (Gall et al., 1992), sensitive neurons of the neural crest, and placodes. It is crucial for the survival of sympathetic and sensitive neurons, which are later supported by NGF and BDNF (Fariñas et al., 1994). Nt-3 is involved in the processes of neurogenesis, hippocampal plasticity, learning and memory (Shimazu et al., 2006). An increase in NT-3 levels in the serum of schizophrenia patients with predominantly negative symptoms has previously been reported but the underlying mechanism remains unclear (Arabska et al., 2018). Some polymorphisms of the NTF3 gene turned out to be associated with the incidence of psychiatric and cognitive disorders, although they appear to have different significance in different populations. For example, a dinucleotide polymorphism (CA23) in the A3/147 bp NTF3 promoter region was found to be associated with schizophrenia in a Japanese population (Nanko et al., 1994). However, these data have not been confirmed in other studies in American and European Caucasian populations (Dawson et al., 1995; Nimgaonkar et al., 1995; Jŏnsson et al., 1997; Virgos et al., 2001)—statistically significant correlations with schizophrenia predisposition have only been observed in females (Virgos et al., 2001). A statistically significant association between the frequency of NTF3 gene SNP rs1805149-A (G76E) and the incidence of schizophrenia was found only in patients with the early manifestation (earlier than 25 years-old) and patients with a significant duration of the disease (more than 10 years) (Hattori and Nanko, 1995). For NTF3 rs6489630-T (3′ downstream) a statistically significant association was established with lower intelligence scores in children with ADHD (Cho et al., 2010) and the predisposition to gambling disorder (haplotype with rs7956189-G [3′ downstream)] was found (Solé-Morata et al., 2022). Despite the importance of NT-3 in brain morphogenesis, the association of genomic variants of NTF3 with predisposition to mental and cognitive disorders only in limited and narrow selections, which leaves the role of NTF3 SNPs in psychiatric diseases in question.
As for the NT-3 TrkC receptor (NTRK3 gene), some of its genomic variants were identified in patients with mental disorders. In particular, NTRK3 SNPs rs12595249-C (intr), rs744994-T (promoter), and rs998636-G (promoter) was found to be associated with the predisposition to drug addiction in a Spanish population (Fernàndez-Castillo et al., 2013), C/T heterozygote in rs7180942 (intr) SNP—with the predisposition to eating disorders (Mercader et al., 2008), rs8037291-G (intr) variant—with the occurrence of ADHD (Smith et al., 2014) and rs1946698-C (intr)—with the predisposition to schizophrenia in a Russian population (Lu et al., 2013). However, functional significance of these SNPs in psychiatric and cognitive disorders remains to be elucidated.
Neurotrophin-4 (NT-4, NTF4 gene) is expressed predominantly in dopaminergic, GABAergic neurons of the ventral midbrain and hippocampal neurons; it is also found in the thalamus, hypothalamus, medulla oblongata, cerebellum and pontine (Holtzman and Mobley, 1994). NT-4 promotes proliferation, differentiation, and survival of neural crest and placode sensory neurons (Ip et al., 1992), motoneurons, neurons of the basal forebrain and locus coeruleus (Friedman et al., 1993), and stimulates synaptic activity in hippocampal cultures in vitro (Friedman et al., 2002). Similarly to BDNF, TrkB and p75NTR function as receptors for NT-4. Since NT-4 participates in the processes of synaptogenesis, neuronal survival and neural plasticity, it may take part in the pathogenesis of psychiatric and cognitive disorders, like other neurotrophins. However, this assumption has not yet been experimentally confirmed—only an increase in NT-4 serum concentration was established in bipolar disorder (Loch et al., 2015), but not in schizophrenia (Skibinska et al., 2019). Kranz et al. (2015) also detected a genomic variant rs746640305-A (D162Y) in the NTF4 gene in one of 48 patients with schizophrenia-related psychosis (American population); however, its functional significance was not experimentally verified. To date, there are no data on the association of certain genomic variants of NTF4 with the predisposition to mental or cognitive disorders.
3.3 Glial neurotrophic factors and their receptorsGlial cell line-derived neurotrophic factor (GDNF, GDNF gene) is an ancestor to the family of glial neurotrophic factors, which includes neurturin (NRTN), artemin (ARTN), and persephin (PSPN) along with GDNF (Airaksinen and Saarma, 2002). GDNF is involved in the development, maintenance, survival, and function of dopaminergic and motor neurons in the mammalian nervous system (Lin et al., 1993; Airaksinen and Saarma, 2002). The GDNF gene is localized in the 5p12—p13.1 locus associated with schizophrenia (Moises et al., 2002; Bespalova et al., 2005).
Given its broad functional in the nervous system, GDNF has been considered to potentially contribute to the pathogenesis of mental diseases, including schizophrenia, known for dopaminergic system dysfunction. There is indirect evidence for such a link: particularly, application of drugs that cause schizophrenia-like symptoms elevates GDNF expression. For example, phencyclidine increases the GDNF expression, as well as of its receptors, GFRα1 and RET (Semba et al., 2004). Amphetamine administration elevates the endogenous GDNF expression in the nigrostriatal tract (Morrow et al., 2011; Valian et al., 2017), increases the concentration of dopamine in synapses, and elevates the schizophrenia susceptibility (Morrow et al., 2011; Li et al., 2014; Valian et al., 2017). It has also been demonstrated that a 2–3-fold increase in endogenous GDNF expression triggers schizophrenia-like conditions (including the expression of relevant genes and behavioral responses) and also imbalance the dopamine content between the prefrontal cortex and striatum (Ross et al., 2008; Liao et al., 2012; Wearne and Cornish, 2018).
At the same time, only a small number of studies have been devoted to investigating the relationship between the occurrence of GDNF genomic variants and the predisposition to psychiatric disorders (Lee et al., 2001; Ma et al., 2013), with no reliable data obtained to definitively confirm such correlations. For example, no reliable associations were found between the frequency of trinucleotide repeats (AGG)n (and a number of GDNF missense mutations) and the incidence of psychiatric disorders in a Japanese population. In contrast, a polymorphism containing 15 or more AGG repeats was identified as a protective factor in an Italian population (Michelato et al., 2004). Studies in English and Chinese populations also revealed no functional significance of GDNF polymorphisms: (AGG)n, rs2910709-C/G/T (3′ downstream), rs2973050-C/G/T (intr), rs884344-C (intr), rs2910702-A/T (intr), rs2216710-T (intr), rs3812047-G/T (intr), etc. in the development of mental disorders (Kotyuk et al., 2013).
GDNF signaling is mediated by the heterotetrameric receptor complex, consisting of GFRα1 (GDNF family receptor alpha-1) and RET (Receptor Tyrosine Kinase) (Airaksinen and Saarma, 2002). Abnormalities in the structure and signaling of GFRα1 (GFRA1 gene) or RET (RET gene), caused by genomic variants or mutations within these genes, may also underlie various mental and cognitive disorders. For example, an association was determined between the incidence of rs11197557-T (intr) SNP in the GFRA1 gene and predisposition to schizophrenia (Souza et al., 2010). Interestingly, this polymorphism is localized in the locus of chromosome 10, which is associated with schizophrenia predisposition.
Impaired expression of artemin (ARTN gene) results in altered nervous system development (Artn knockout) in a murine model (Honma et al., 2002) or in the increased excitability of striatal dopamine neurons in humans (Elitt et al., 2006, 2008). rs11242417-G (intr) polymorphism in the artemin receptor GFRα3 gene (GFRA3) turned out to be associated with the schizophrenia predisposition (Souza et al., 2010).
No genomic variants for RET (the signal-transducing subunit of heterotetrameric neurotrophin receptor from the GDNF family) have been identified in association with the susceptibility to psychiatric disorders. At the same time, a complete deletion of RET activity coinciding with schizophrenia manifestation in a European population have been elucidated (Glessner et al., 2010).
3.4 Cytokines and their receptors. IL-6Ciliary neurotrophic factor (CNTF, CNTF gene), a member of the neuropoietic cytokine family, is expressed in the brain and spinal cord (Pasquin et al., 2015). CNTF supports the survival and proliferation of hippocampal neurons, medial septal neurons, and appears to be involved in development of the central nervous system (Sakai et al., 1997).
The role of CNTF in the pathogenesis of psychiatric disorders has not been elucidated. Most studies indicate that there is no correlation between the incidence of psychiatric disorders and the prevalence of known CNTF genomic variants (Sakai et al., 1997; Tanaka et al., 1998; Benkovits et al., 2016). For rs1800169 (FS63Stop) a correlation with the incidence of schizoaffective disorder and a better response to iloperidone (antipsychotic) therapy was reported (Lavedan et al., 2008; Okahisa et al., 2010). A number of studies demonstrate that impaired CNTF signaling caused by mutations in the genes of the receptor complex CNTFR*gp130 (CNTFR and IL6ST genes, respectively) may contribute to mental disorders. Moreover, some SNPs in the CNTFR gene have been associated with the predisposition to drug addic
留言 (0)