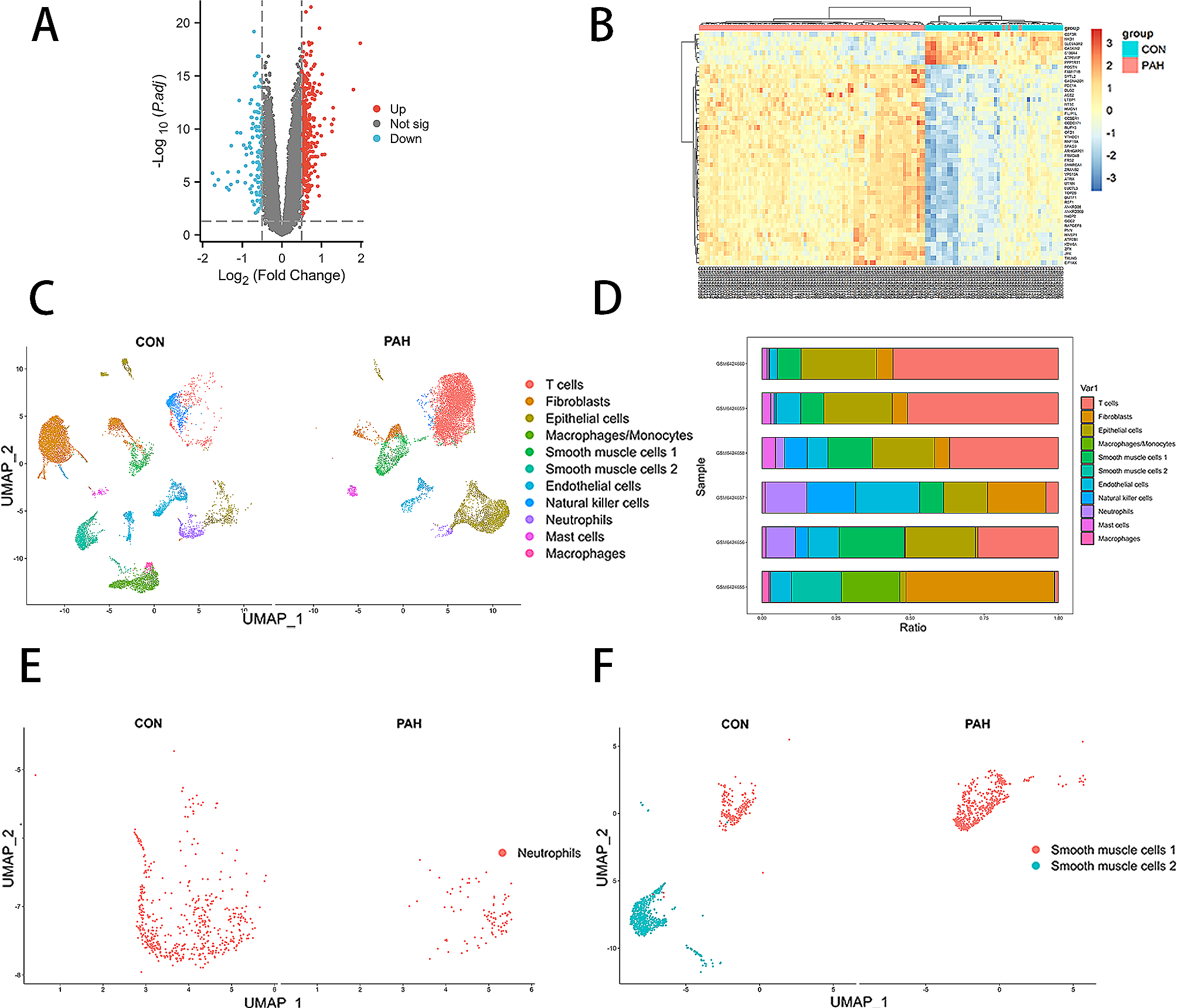
Cell culture and treatment
The A549 cell line, MLE-12 cell line, BEAS2B cell line, and MRC-5 cell line were all purchased from American Type Culture Collection and cultured in a standard medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/L streptomycin as previously described. All cell lines were tested and confirmed to be mycoplasma-free.
Plasmids and transfection
The human NR2F2 and mouse Nr2f2-overexpressing pcDNA3.1 plasmid or the empty pcDNA3.1 plasmid were synthesized and transfected into A549, BEAS2B and MLE-12 cells using Lipofectamine 3000 according to the manufacturer’s protocol. Human NR2F2 and mouse Nr2f2 shRNAs were designed and synthesized by Sangon Biotech and subsequently annealed and inserted into the pLKO.1 vector (Sangon, Shanghai, China).
Western blot (WB) analysis
Western blot analysis was performed following previously described methods [25]. Briefly, lung tissues and cells were lysed in the RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) supplied with protease and phosphatase inhibitors. The protein concentration of the supernatant was quantified by the BCA kit (Solarbio, Beijing, China). Equal amounts of protein were separated on 8–12% SDS-PAGE gels and then transferred onto PVDF membranes (Millipore, Darmstadt, Germany). After blocking with 5% skim milk, membranes were incubated with specific primary antibodies overnight at 4 °C. The membranes were then incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Blots were visualized on the LI-COR Odyssey Fc device (LI-COR Biosciences; Lincoln, NE).
Quantitative real-time PCR (qRT‒PCR)
For transcriptional analysis of tissue samples or cultured cells, total RNA was extracted using TRIzol (Takara, Dalian, China) as previously described [26]. cDNA was transcribed using the GoScript™ Reverse Transcription System (Promega Corporation, Wisconsin, USA). Afterward, qRT‒PCR were performed using Light Cycler 480 fluorescent quantitative PCR system (Roche) and SYBR green real-time PCR master mix (QIAGEN, Hilden, Germany) with cDNA as the template. The fold change of gene expression was calculated as 2 − ΔΔCt. The primer pairs used in this study are described in Table 1.
Table 1 Genes selected for expression analysisCell counting kit-8 (CCK8) assay
Cell viability was assessed by a CCK8 assay following the manufacturer’s instructions. In short, A549 cells were seeded at a density of 3 × 103 cells/well in 96-well microplates. After indicated transfection for 48 h, 10 μL of CCK-8 solution was added to each well and incubated at 37 °C for 2 h. The absorbance was then measured at 450 nm using a microplate reader.
EdU assay
Cell proliferation was measured by an EdU assay kit (RiboBio, Guangzhou, China). A549 cells were seeded in 96-well plates at 3 × 103 cells per well. After transfection for 48 h, each well was incubated with 50 μM EdU medium for 2 h at 37 °C. The cells were then fixed in 4% paraformaldehyde for 30 min and permeabilized with 0.5% Triton X-100 for 10 min. After PBS washes, the cells were incubated with 1×Apollo reaction cocktail for 30 min. Subsequently, 1×Hoechst 33,342 was added for 30 min. The stained cells were observed and imaged using a fluorescence microscopy (Leica, Wetzlar, Germany).
Transwell assay
The treated MRC-5 cells were digested and approximately 2 × 104 cells in 100 μL serum-free DMEM were placed in the upper transwell chamber, and 600 μL medium containing 10% fetal bovine serum was added to the lower transwell chamber. The 24-well plate was incubated at 37 °C for 24 h. After 24 h incubation, the cells were fixed with 4% paraformaldehyde for 30 min, stained with crystal violet solution for 10 min, observed under electron microscope and imaged.
Senescence-associated β-galactosidase (SA-β-gal) staining
The activity of SA-β-gal was performed by a senescence β-galactosidase staining kit following the manufacturer’s instructions (Solarbio, Beijing). Briefly, the treated cells on 24-well chamber slides and lung tissues were fixed with 4% formaldehyde for 15 min at room temperature, rinsed three times with PBS for 3 min, and then incubated with freshly prepared SA-β-Gal staining solution at 37 °C overnight. Slides were rinsed twice with PBS for 1 min at room temperature. The positive cells were observed and imaged using an electron microscope.
Collagen gel contraction assay
Lung fibroblast contraction assay was conducted using a two-step cell contraction assay kit following the manufacturer’s guidelines (CBA-201, Cell Biolabs, Inc, San Diego, CA) as previously described [26]. The area of gel contraction was measured using ImageJ software (ImageJ 1.52q). The gel contraction rate was calculated as the contracted area divided by the initial gel release surface area.
Mouse model of bleomycin-induced pulmonary fibrosis and IPF
The animal maintenance and handling procedures followed the Henan Normal University Institutional Animal Care and Use Committee (IACUC, SMKX-2118BS1018) guidelines, which coordinate with the Association of Animal Behavior and National Regulations. Eight-to-ten-week-old C57BL/6 N male mice were purchased from Beijing Charles River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained in a specific pathogen-free environment. For bleomycin-induced PF, a single 50 μl injection containing 1.5 U/kg of bleomycin (Nippon Kayaku Co., Tokyo, Japan) diluted in PBS or PBS only (vehicle) was intratracheally administered. At designated time points, mice were sacrificed by intraperitoneal injection of ethyl carbamate, and samples were collected for further analysis.
IPF lung tissues and control non-IPF lung tissue samples were recruited based on the ATS/ERS/JRS/ALAT Clinical Practice Guidelines at Henan Provincial Chest Hospital. The study was approved by the Henan Provincial Chest Hospital Medical Research Ethics Committee (No. 2019-05-07), and informed consent was obtained from all the patients before surgery. The work was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
Measurement of hydroxyproline
The lung hydroxyproline content was determined using the hydroxyproline colorimetric assay kit (MAK008, Sigma, St. Louis, MO, US) according to the manufacturer’s instructions, as previously described [26]. The results were calculated as μg hydroxyproline per right lung.
Hematoxylin and eosin (H&E) and Masson’s trichrome staining
H&E and Masson’s trichrome staining were conducted following the protocol described in reference [26]. Mouse lung tissues were fixed in 4% paraformaldehyde for 24 h, dehydrated, and embedded in paraffin. Sections (4 μm) were routinely deparaffinized in distilled water. Morphological analysis was performed using a staining kit, following the manufacturer’s instructions, for H&E and Masson’s trichrome staining.
Immunohistochemistry (IHC) and immunocytochemistry (ICC)
IHC was performed as previously described [1]. Briefly, the lung tissue samples were fixed with 4% paraformaldehyde, embedded in paraffin wax, and sectioned into 4 μm sections. The sections were then deparaffinized, rehydrated and treated with endogenous peroxidase blocking solution (Beyotime) for 10 min to quench endogenous peroxidase activity. Subsequently, the lung sections were accomplished with citrate buffer (Beyotime) at 100 °C for 10 min. After washing with PBS, the slides were blocked with blocking solution (Beyotime) at 37 °C for 30 min before overnight incubation with primary antibodies (anti-NR2F2). Biotin-labeled secondary antibodies (Beyotime) were applied at 37 °C for 30 min after washing. Next, the lung sections were developed with DAB working solution, counterstained with hematoxylin. Stained sections were visualized and photographed using light microscopy.
For ICC, cells were cultured on coverslips coated with poly-L-lysine, immobilized by 4% paraformaldehyde for 30 min at room temperature and permeabilized with 0.03% Triton X-100 for 5 min. After rinsing three times with PBS, the cells were blocked with 5% goat serum for 30 min, followed by incubation with primary antibodies (anti-P21, anti-γ-H2AX, anti-ki67) at 4 °C overnight and Alexa Fluor 594 (red)-conjugated secondary antibodies at 37 °C for 1 h and DAPI. The fluorescence was visualized under confocal microscope (LSM 700, Zeiss, Jena, Germany).
Micro-CT imaging
Fourteen days after bleomycin administration, in vivo micro-CT analysis of the entire lung was conducted. Briefly, mice were lightly anesthetized with isoflurane and fixed in the supine position. Micro-CT images were acquired using a Bruker SkyScan 1276 micro-CT system (Bruker, Kontich, Belgium). The scanning parameters were set as follows: X-ray tube voltage of 60 kV and anode current of 200 μA, with a Cu filter of 0.5 mm. The total acquisition time was approximately 10 min. The reconstructed images were superimposed using Insta-Recon software (Bruker microCT, Kontich, Belgium).
ELISA assay
The concentrations of IL-1β (Solarbio, Beijing) in mouse bronchoalveolar lavage fluid (BALF), the concentrations of TGF-β1 (Solarbio, Beijing) in the cell culture medium and the levels of 8-OHdG (Abcam, England) in cells were measured using ELISA assay kits, following the manufacturer’s instructions.
Comet assay
Cellular DNA damage was assessed using the comet assay, following the previously described protocol [27]. In brief, cells were embedded in agarose gel and subjected to electrophoresis to allow the fragmented and denatured DNA strands to migrate, forming a comet-like pattern. Subsequently, ethidium bromide staining was performed, and the samples were observed and photographed under a fluorescence microscope for analysis (Leica, Wetzlar, Germany).
Detection of ROS
The intracellular ROS level was assessed using a ROS assay kit (Applygen Technologies Inc., Beijing, China). Briefly, the treated A549 cells were incubated with 10 μM dihydroethidium at 37 °C for 20 min in the dark. Following incubation, the cells were washed twice with PBS carefully, and the intensity of fluorescence was determined by fluorescence microplate reader at an excitation wave length of 535 nm and emission wave length of 610 nm.
Isolation of primary alveolar epithelial cells (AECIIs)
Primary AECIIs were isolated from the lung tissues of 2-month-old wild-type C57BL/6 mice using the protocol described in the reference [1]. Briefly, 20 mL of wash buffer was perfused into the mouse lungs, followed by the intratracheal injection of 3 mL of collagenase. The lungs were then excised, incubated at 37 °C for 1 h, and subsequently minced using a gentleMACS dissociator. The minced tissue was sequentially filtered through 100, 70, and 40 μm cell strainers, followed by gradient centrifugation (300 g, 20 min). The intermediate cells were resuspended in BEGM medium and cultured in dishes precoated with CD45/32/16. Using this protocol, AECIIs with over 80% purity were obtained and used for subsequent experiments.
Statistical analyses
Statistical analyses were performed by GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA). The Shapiro-Wilk normality test was employed to assess normal distribution. For non-normally distributed data, the Mann-Whitney U test was used to compare two groups, while the unpaired Student’s t-test was utilized for comparisons between two groups with normally distributed data. All data are shown as the mean ± standard deviation (SD) and were considered statistically significant at P < 0.05.





留言 (0)