
記住我


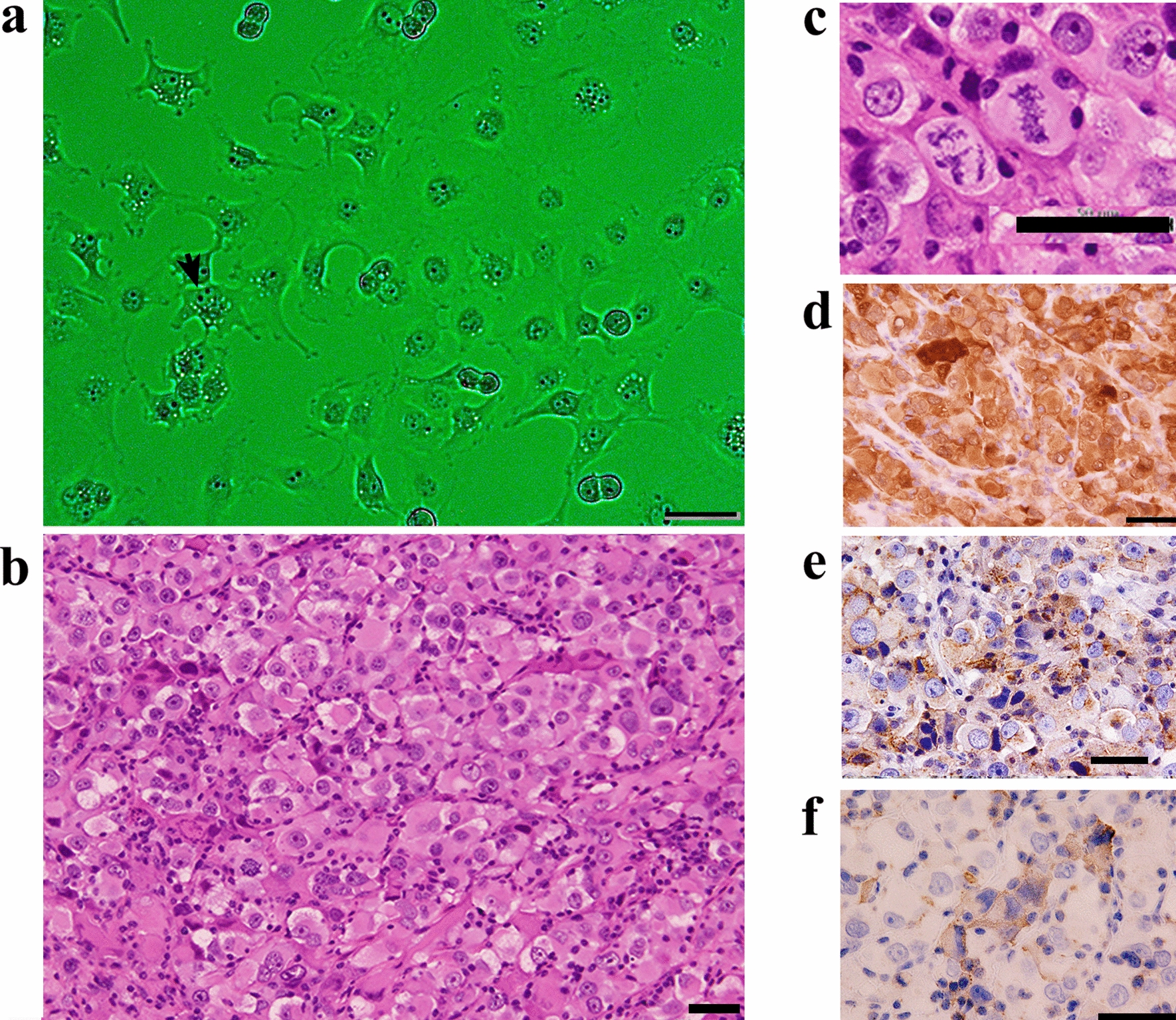
A 46-year-old Japanese woman, gravida 0, was referred to the hospital with complaints of anorexia and abdominal pain. Systemic computed tomography (CT) and abdominal magnetic resonance imaging (MRI) revealed an 11 cm-sized left ovarian cystic tumour with solid lesions, along with enlarged para-aortic and pelvic lymph nodes, omental mass, metastases to the left lung and left adrenal gland, and massive ascites. Serum cancer antigen 125 (CA125) levels were 4,987 U/mL. These findings overall strongly suggested advanced-stage ovarian cancer, and a staging laparoscopy was performed to assess the extent of peritoneal seeding and to biopsy tumour tissue for pathological diagnosis. Consequently, the predictive index value, a score that predicts surgical outcomes in patients with advanced ovarian carcinoma (ranging from 0 to 14) [9], was found to be 8 for this patient, and the pathological diagnosis of the tumour tissue was clear cell carcinoma (Fig. 1; see Results). The patient was given 3 cycles of intravenous paclitaxel (175 mg/m2) and carboplatin (area under the concentration–time curve [AUC] of 6 mg∙min/mL) on a 21-day cycle. Post-chemotherapy systemic CT revealed progressive disease (PD) according to the Response Evaluation Criteria in Solid Tumors (RECIST). Intravenous pegylated liposomal doxorubicin (50 mg/m2) was then administered as adjuvant chemotherapy. During this time, next-generation sequencing (FoundationOne® CDx) of the patient’s tumour tissue identified multiple pathogenic variants, including ARID1A (Q1346*) and PIK3CB (E1051K), and based on high tumour mutational burden (MTB), pembrolizumab was identified as a potential treatment option. However, the patient suffered acute post-renal failure and sepsis due to increased peritoneal dissemination and died four months after diagnosis of ovarian cancer.
Fig. 1
Histological appearance of the patient’s tumour. Tissue sections were stained with hematoxylin and eosin (H&E). Arrowheads in right panel indicate hyaline bodies with a target-like or bull’s-eye appearance. Bars = 50 μm, left panel; 20 μm, right panel
Isolation of carcinoma cellsA fragment of ovarian cancer tissue obtained at the time of staging laparoscopy was cut into small pieces, suspended in 10 mL of 1 mg/mL Collagenase/Dispase (Roche Diagnostics, Mannheim, Germany) and incubated at 37 °C for 30 min [10]. The slurry was passed through a 40-μm pore cell strainer to remove undigested tissue fragments. After washing with phosphate-buffered saline (PBS), cells were cultured in RPMI 1640 medium supplemented with 10% FBS (HyClone, South Logan, UT), 100 U/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B (Nacalai Tesque, Kyoto, Japan). Two weeks later, we observed a colony of neoplastic epithelial cells from which the cell line MTC-22 was cloned using stainless steel cloning cylinders and trypsin. Growth and morphology of cells in culture were observed under an inverted phase-contrast microscope IX71 (Olympus, Tokyo, Japan). Use of fresh human ovarian cancer tissues was approved by the Research Ethics Committee of University of Fukui (reference number 20200176, approved on February 22, 2021) and written informed consent was obtained from the patient.
Transmission electron microscopyCells grown to confluency were pre-fixed in 2% paraformaldehyde/2% glutaraldehyde in 30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4) for 30 min. After washing with HEPES buffer, cells were post-fixed with 1% osmium tetroxide in HEPES buffer at 4 °C for 30 min. After washing with 10% sucrose in distilled water, cells were stained with 2% uranyl acetate for 60 min. After dehydration in a graded series of ethanol, cells were embedded in epoxy resin (Epon 812) (Nisshin EM, Tokyo, Japan). Ultrathin sections were stained with uranyl acetate and lead citrate, and observed under an H-7650 transmission electron microscope (Hitachi High-Tech, Tokyo, Japan).
Growth curve analysis and doubling-timeCells were seeded into wells of 6-well plates at a concentration of 1.0 × 104 cells/well. The number of cells per well was determined in triplicate at 24-h intervals over an 8-day period, and a growth curve was drawn. Doubling-time was calculated using a web-based calculator (https://www.doubling-time.com/compute.php).
Cell cycle analysisCells were trypsinized to establish single cells and fixed in ice-cold 70% ethanol at 4 °C for 2 h. Cells were then re-suspended in 0.5 mL PBS, and 5 μL Cell Cycle Assay Solution Blue (Dojindo Laboratories, Mashiki, Japan) was added and incubated 15 min at 37 °C under light-shielded conditions. Stained nuclei were analysed using FACSCanto II (BD Biosciences, San Jose, CA) with FlowJo software (Tree Star, Ashland, OR).
KaryotypingCells were harvested after 3 h of colcemid treatment to arrest cells in metaphase, treated with hypotonic solution (75 mM potassium chloride) for 20 min, and fixed with Carnoy’s solution (3:1 ratio of methanol and acetic acid). Cell suspensions were dropped onto glass slides and chromosome spreads were prepared using a HANABI Metaphase Spreader (ADSTEC, Funabashi, Japan). Slides were dried 18 h at room temperature, stained with Giemsa solution, and mounted with coverslips. Metaphase spreads were captured using an Axio Imager Z2 microscope (Carl Zeiss, Oberkochen, Germany) and analysed using Ikaros karyotyping software (Metasystems, Altlussheim, Germany). Karyotypes constructed from G-banded chromosomes were described according to the International System for Human Cytogenomic Nomenclature (ISCN) 2020 [11].
Sanger sequencingARID1A and PIK3CB mutations in MTC-22 cells were detected by Sanger sequencing. Total RNA was extracted from cells using ISOGEN reagent (Nippon Gene, Tokyo, Japan) as per the manufacturer’s protocol, and single-stranded cDNA was synthesised as described [12]. ARID1A DNA fragments corresponding to amino acid residues 1262–1428 and PIK3CB fragments corresponding to amino acid residues 967–1133 were amplified by polymerase chain reaction (PCR) using PrimeSTAR® MAX DNA Polymerase (Takara Bio, Kusatsu, Japan) with the following oligonucleotide pairs: 5'-ACCCAAgCTggCTAgCTgCTgCCggCCCTgggCT-3' and 5'-gCCCTCTAgACTCgAgTgTATACATCTTgCTgAggg-3' for ARID1A; and 5'-ACCCAAgCTggCTAgCCATTCAACAAggAAAAACAgg-3’ and 5'-gCCCTCTAgACTCgAgAAgCAgAgggAATCATCgg-3' for PIK3CB. Resultant PCR products were subjected to 1% agarose gel electrophoresis, and bands of the expected size (~ 500 bp) were cut from the gel and purified using QIAquick® Gel Extraction Kit (QIAGEN, Venlo, The Netherlands). Purified DNA fragments were inserted into NheI/XhoI sites of pcDNA3.1 using In-Fusion Snap Assembly Master Mix (Takara Bio). Sequencing reactions were carried out using a BigDye® Terminator v.1.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA) as per the manufacturer’s protocol, and sequenced using an Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific).
Short tandem repeat (STR) analysisGenomic DNA was extracted from cells using a NucleoSpin® Tissue kit (Takara Bio), and 16 STR loci were detected by multiplex PCR using a PowerPlex® 16 HS System (Promega, Madison, WI). STR profiles were compared with those recorded in the Expasy Profile Database (https://www.cellosaurus.org/str-search/), as described [13].
Monoclonal antibodiesThe following monoclonal antibodies served as primary antibodies: BC12 (mouse IgG; Nichirei Biosciences, Tokyo, Japan) recognising paired box 8 (PAX8); EPR18644-13 (rabbit IgG; Abcam, Cambridge, UK) recognising hepatocyte nuclear factor 1β (HNF1β); WT49 (mouse IgG; Leica Biosystems, Newcastle Upon Tyne, UK) recognising Wilms tumour 1 (WT1); DO-7 (mouse IgG; Nichirei Biosciences) recognising p53; 1D5 (mouse IgG; Dako, Glostrup, Denmark) recognising oestrogen receptor (ER); 1A6 (mouse IgG; Dako) recognising progesterone receptor (PgR); OV-TL 12/30 (mouse IgG; Dako) recognising cytokeratin 7 (CK7); Ks20.8 (mouse IgG; Dako) recognising CK20; V9 (mouse IgG; Dako) recognising vimentin; GP1.4 (mouse IgG; Leica Biosystems) recognising epithelial membrane antigen (EMA); Ov185:1 (mouse IgG; Leica Biosystems) recognising CA125; R-10G (mouse IgG; Tokyo Chemical Industry, Tokyo, Japan) [14, 15] and 294-1B1 (mouse IgM) [16], both recognising low-sulphated keratan sulphate; and 5D4 (mouse IgG; Seikagaku, Tokyo, Japan) recognising highly sulphated keratan sulphate.
Histological and immunohistochemical analysisFormalin-fixed, paraffin-embedded tissue sections were stained with hematoxylin and eosin (H&E) or immunostained with the monoclonal antibodies noted above. Immunohistochemical staining was undertaken using the Histofine system (Nichirei Biosciences), according to the manufacturer’s protocol. Analysis of human ovarian cancer tissues was approved by the Research Ethics Committee of University of Fukui (reference number 20200024, approved on May 19, 2020).
Immunofluorescence stainingImmunofluorescence staining of MTC-22 cells was performed essentially as previously described [17, 18]. Briefly, cells grown on coverslips were fixed 15 min with neutralised 20% formalin (pH 7.4). After permeabilizing the cell membrane with 1% Triton X-100 in PBS for 15 min, cells were incubated 30 min with the primary antibodies noted above, followed by a 15-min incubation with Alexa Fluor 488-conjugated species- and class-matched secondary antibodies (Thermo Fisher Scientific) supplemented with 4'6-diamidino-2-phenylindole (DAPI). Cells were mounted in 50% glycerol in Tris-buffered saline (TBS) and observed under an AX-80 fluorescence microscope (Olympus).
留言 (0)