
記住我



Enterococci are gram-positive, chain-forming, non-spore-forming, facultative anaerobic lactic acid bacteria (LAB), commonly isolated from the gastrointestinal tract (GIT) of humans and animals [3]. They delineated from Vagococcus ~ 500 million years ago, have co-evolved with animal territorialisation and have associated heavily with the mammalian GIT [4]. The human gut microbiota hosts approximately < 0.1% enterococci [4]. Previously classified as part of the group D Streptococcus based on the Lancefield serologic typing system, they were acknowledged as a separate genus in the 1980s, Enterococcus [5]. This genus currently contains 83 species [6]. Comparative genomic analysis of 37 Enterococcus strains revealed that this genus represents a group with variation in GC content (34–45%) and genome size (2.31 Mbp to 5.5 Mbp) [4, 7]. Functional analysis of the pan-genome highlights the flux of niche-specific genes (NSG) over time, where the greatest flux of annotatable genes is associated with carbon utilisation, phosphotransferase systems (PTS) and transcriptional regulation [4]. This indicates the evolution of Enterococcus coupled with horizontal gene transfer (HGT) events, selective pressure, and niche transition. Enrichment of genes involved in cell wall modification, de novo purine biosynthesis and stress response suggests adaptability to niche diversification and phenotypic resilience due to genomic plasticity resulting in genus diversification [4].
The genus, previously as part of Streptococcus, was first described as “hardy” in 1899. An analysis of phenotypic growth in the presence of stressors associated with the hospital environment found ubiquitous resistance to β-lactam antibiotics and common disinfectants. E. faecium and E. faecalis were some of the most desiccant and starvation resistant, respectively [4, 8]. The enterococcal pangenome contains ~ 29,545 gene families and grows continuously, pointing to an open pangenome suggesting gene exchange within and between species [7]. However, this is not the case for all species within the genus, as E. faecium and E. faecalis have open and closed pan-genomes, respectively [9, 10]. Habitats drive the evolution of Enterococcus, and genetic relationships are more similar in strains that come from the same environment [11]. Phylogenetics of the core-genome show that human and mammalian isolates are dispersed in branches of E. faecium, E. dispar and E. pallens, while plant and bird isolates are mainly in the E. casseliflavus branch suggesting dissemination of the genus among mammals [7].
Enterococci are used in the fermentation of certain types of cheeses (e.g. traditional European cheeses) and meat products (e.g. fermented sausage) [12]. They are also causative agents of food spoilage, mainly of cooked meats [13]. Enterococci have also been successfully used as probiotics but remain controversial for such applications given their genetic promiscuity and relatedness to pathogenic strains [13, 14]. They are part of the commensal microbiota in the gut but can cause infectious diseases, such as endocarditis, urinary tract infections (UTI) and bacteraemia. The two species most associated with invasive infection are E. faecium and E. faecalis. Treatment of the resulting diseases is often complex due to their resistance to commonly used chemotherapeutic agents [5]. The first documented use of the term “enterococcus” in 1899 highlighted the bacterium’s ability to become pathogenic; presently, E. faecium represents a pathobiont currently a threat to global health [15]. One of the “hottest” issues regarding pathogenic enterococci is the emergence of multidrug resistant (MDR) strains, leading to enterococci becoming the 2nd most causative agent of hospital-acquired infections (HAI) [1]. Enterococci also enhance the pathogenesis of Clostridioides difficile suggesting their role in poly microbial infections [16].
On average, E. faecium clinical isolates (CL) harbour 10 resistance genes, including vancomycin, aminoglycoside, macrolide-lincosamide-streptogramin, and tetracycline [17]. Daptomycin, a first-line treatment for VRE, was recently shown to select for off-target resistance within the human after intravenous treatment [18]. Non-synonymous mutations conferring resistance to daptomycin are detected globally, indicating the emergence of resistant mutants due to local selective pressures. However, these do not correlate significantly with vancomycin resistance genes [17]. Evidence is also emerging of small colony variants (SCV) among E. faecium and E. faecalis species, a phenomenon usually associated with increased robustness, antibiotic resistance and recurrent infections. To date, described cases of SCVs among enterococci are vancomycin susceptible [19, 20].
This review provides detail on the mechanisms of vancomycin resistance in enterococci. We examine the evolutionary relationships between hospital-associated pathogenic enterococci and their community counterparts based on genomics and present the likely routes of transmission based on this data. Finally, we look at conventional and novel approaches for treating VRE infections, including antibiotics and combinations thereof, non-antimicrobial-based drugs, bacteriocins, bacteriophage therapy, probiotics, vaccines and the commensal gut microbiota itself.
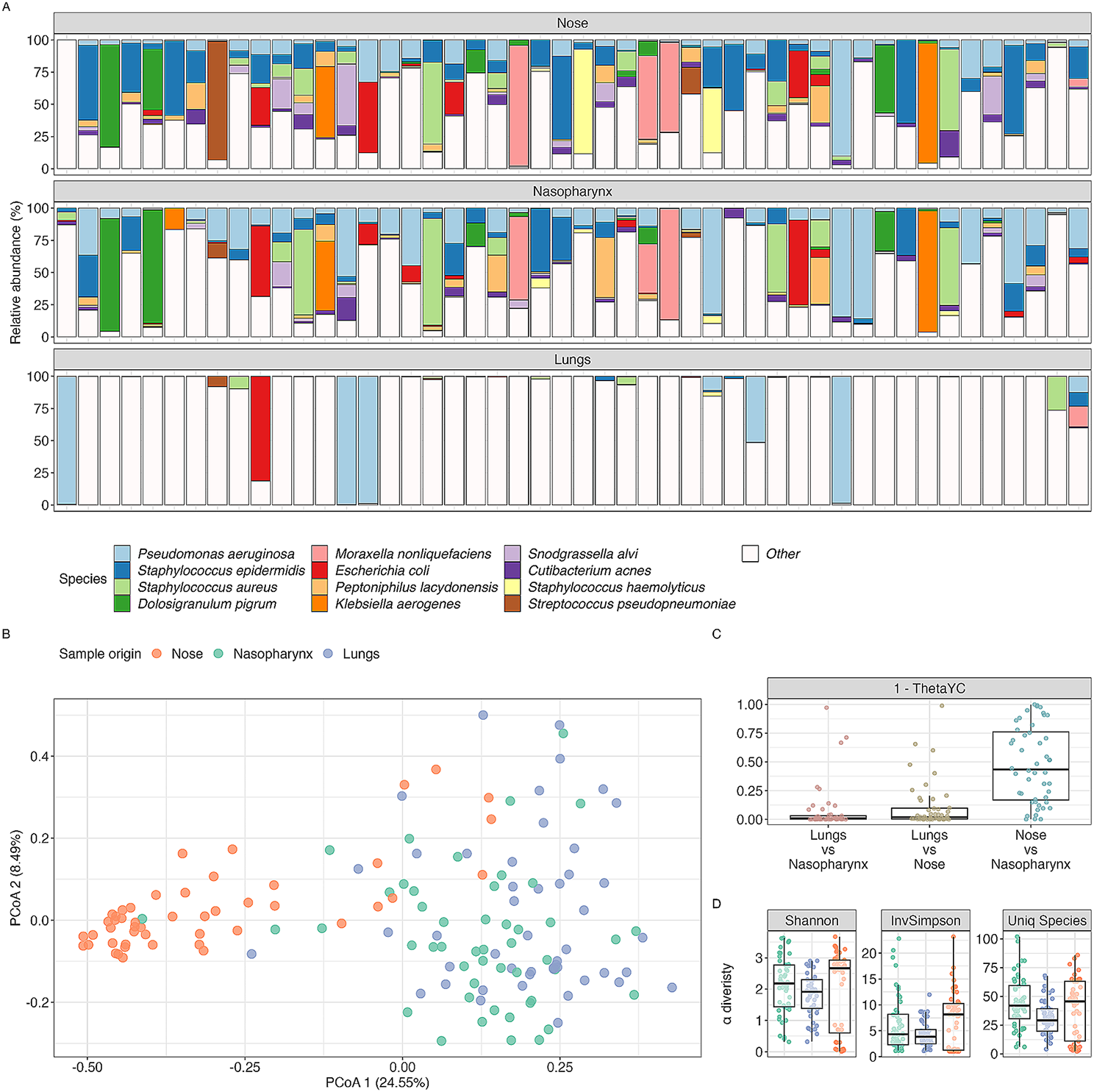
Vancomycin resistance in enterococciEnterococci possess intrinsic resistance to several groups of antibiotics, such as tobramycin, kanamycin, β-lactams and lincosamides (clindamycin, streptogramin) [21]. Due to their genome plasticity, enterococci quickly adapt to environmental changes [22]. HGT enables the acquisition of genetic elements that provide resistance to antibiotics and enable survival and persistence of enterococci in clinical settings (Fig. 1). E. faecium and E. faecalis represent two of the hardiest enterococcal species with capabilities to withstand multiple antibiotics, antiseptics, salt concentrations, organic compounds such as sorbic acid, and other stressors such as urea and high pH [4].
Fig. 1
a Enterococcal acquisition of niche specific genes and (b) dissemination routes for VanA genes. a A change of niche resulted in Enterococcus spp. acquiring a harder cell wall structure and increased mutatable phenotype. E. faecium acquired various substrate utilisation genes, namely glucose, mannose, galactose and fructose. Hospital acquired infection-related E. faecium lack CRISPR-Cas systems, rendering them susceptible to receiving ectopic DNA resulting in the acquisition of pathogenicity islands, plasmids and insertion sequences. Hospital-acquired, hypermutable clade E. faecium can also have single nucleotide polymorphism (SNP) mediated resistance to antibiotics, such as fosfomycin, an antibiotic used to treat acute non-complicated urinary tract infections (UTIs). A hypermu table phenotype due to mutations in the DNA-mismatch repair proteins MutS and MutL increases the mutation frequency of strains. Enterococci act as genomic reservoirs for antimicrobial resistance (AMR) genes which can then be passed to recipients like S. aureus and S. gordonii. b The possible dissemination of VRE AMR genes due to transposons, insertion sequences (IS) and plasmids. Nested mobile genetic elements (MGEs) resemble the Russian doll model similar to carbapenemase resistance genes in Enterobacteriaceae, resulting in numerous horizontal dissemination routes including movement of the plasmid, transposition of the transposon between plasmids and homologous recombination [23]. Vertical dissemination occurs through daughter progeny containing the plasmid; this is confirmed by detecting the same plasmids and MGEs amongst the same clonal background. These are often responsible for hospital outbreaks accounting for ~ 30% of dissemination [23]. Horizontal dissemination: (1) Mobilisation of a plasmid to previously susceptible strains via conjugation ~ 7%. (2) Transposon-mediated mobilisation of sequences to other plasmids containing target sequences. (3) Mobilisation of IS. Most cases are caused by separate events indicating the high frequency that strains become pathogenic post-antibiotic treatment [23]. The notation “+” indicates acquisition of DNA, “*” indicates a mutation in DNA, “−“ indicates missing DNA feature
Vancomycin inhibits the formation of the cell wall by binding to the terminal D-Ala-D-Ala dipeptide of cell wall precursors, thus impeding processing into peptidoglycan [24]. Vancomycin bacteriostatic activity against Enterococcus is slow-acting, increasing the potential for the development of resistance [25]. Intrinsically resistant microorganisms possess a naturally different pentapeptide, such as in the case of Lacticaseiobacillus paracasei. Conversely, acquired resistance, as observed in Enterococcus, enables cells to synthesise modified cell wall precursors, for example, replacing the terminal D-ala of the dipeptide with D-lac or D-ser. This structural change results in up to 1000 times lower affinity for binding vancomycin [21].
Vancomycin resistance in enterococci was first described in 1988 [26]. E. faecalis was the predominant source of VRE; however, a gradual transition has occurred over the previous 20–30 years resulting in a species shift of VRE to E. faecium, although resistance is also detected in other enterococci [21, 27]. Vancomycin-resistance genes likely originate from un-sequenced soil bacterial species such as actinomycetes, the natural producers of glycopeptides [28,29,30]. Extensive genomic data from the human gut and skin microbiome suggests that the origin of vanA vancomycin resistance genes lay elsewhere and have moved by HGT, whereas vanB and vanD can be found in gut isolates, notably vanD protein orthologs in the gut commensals Lachnospiraceae and Oscillospiraceae [31, 32].
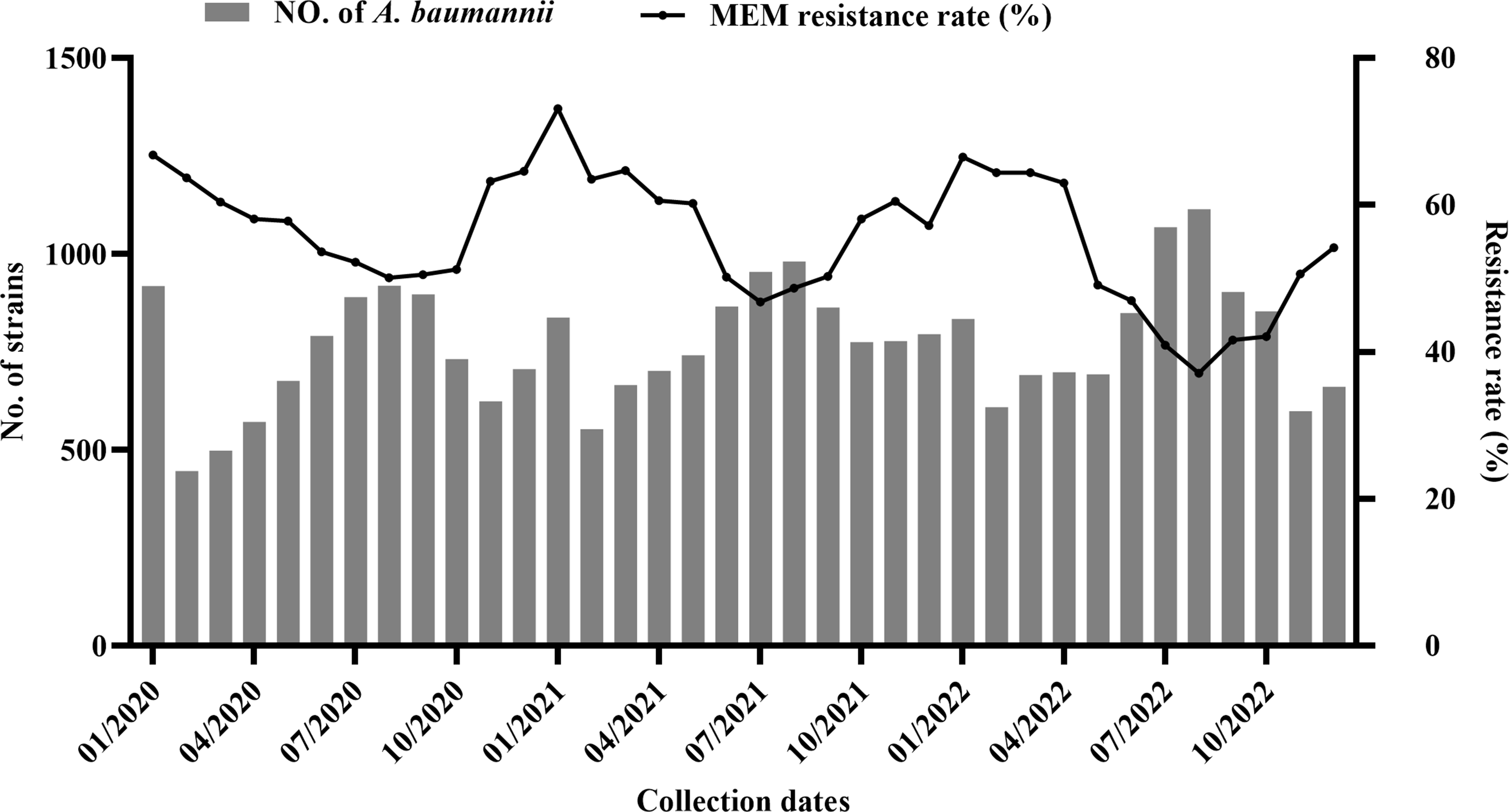
In total, 12 types of vancomycin resistance mechanisms are known, 10 of which are described in enterococci (Fig. 2) [33,34,35,36,37,38]. Two major groups exist, categorised according to ligase activity, which is responsible for replacing the terminal D-ala with D-lac, referred to as D-lac ligases, or D-ser, referred to as D-ser ligases. The operons that encode D-lac ligases often result in high-level resistance with minimal inhibitory concentrations (MICs) > 256 μg/mL (vanA, vanB, vanD and vanM, vanP, vanO, vanI), while operons that encode D-ser ligases result in low-level resistance with MICs of 8–16 μg/mL (vanC, vanE, vanG, vanL, vanN) [21]. Strains of E. gallinarum and E. casseliflavus harbour the vanC operon on their chromosomes, contributing to low intrinsic resistance. E. faecium resistance is conferred by vanA or vanB operons, frequently carried on the transposable elements (TEs) Tn1546 and Tn1549, respectively [39]. HGT of vancomycin resistance has been confirmed among enterococci and other gram-positive bacteria, such as S. aureus, via plasmid transfer. This transfer of resistance is a significant problem, as vancomycin is a last-line antibiotic to treat the rising number of Methicillin-resistant Staphylococcus aureus (MRSA) infections [40].
Fig. 2
Phylogenetic tree of D-Ala-D-(X) ligases. Phylogenetic tree of ligases, those highlighted in blue are present among Enterococcus. The following accessions were used to construct the tree VanA [Enterococcus faecium] (AAA65956.1), VanD [Enterococcus faecium] (AAM09849.1), VanM [Enterococcus faecium] (ACL82961.1), VanC2 [Enterococcus casseliflavus] (AAA60990.1), VanE [Enterococcus faecalis] (ABA71731.1), VanL [Enterococcus faecalis] (ABX54687.1), VanN [Enterococcus faecium] (AEP40500.1), VanF [Paenibacillus popilliae] (WP_006285587.1), D-alanine–D-serine ligase VanG [Clostridioides difficile] (WP_021362548.1), D-alanine–D-serine ligase VanG [Clostridioides difficile] (WP_021425673.1), VanG [Enterococcus faecalis] (AAQ16273.1), D-alanine–D-alanine ligase [Enterococcus faecalis] (WP_002379157.1), D-alanine–D-alanine ligase [Enterococcus faecium] (WP_002293424.1), D-alanyl-alanine synthetase A [Staphylococcus aureus subsp. aureus str. JKD6008] (ADL66141.1), D-alanine–D-alanine ligase [Leuconostoc mesenteroides subsp. mesenteroides J18] (AET29676.1), D-ala D-ala ligase [Lactiplantibacillus plantarum subsp. plantarum ATCC 14917] (EFK27904.1), VanP [Roseburia sp. 499] (WP_075721811.1), VanP [Enterococcus faecium] (WP_222893641.1), VanI [Desulfitobacterium dichloroeliminans] (WP_041219811.1), VanO [Rhodococcus] (WP_209928075.1). The sequences were aligned using muscle [41] and the tree was constructed using RAxML-NG v1.2.0 [42] with 200 bootstrap replicates
Genomics and phylogenetics of disease-related enterococciAlthough E. faecalis is the more common causative agent of enterococcal infections, E. faecium is more intrinsically resistant to antibiotics. Today, more than half of hospital enterococcal isolates in the US are resistant to ampicillin and vancomycin and have high-level resistance to aminoglycosides [22]. The ecological replacement of E. faecalis with E. faecium in the hospital environment could result from the intense use of antibiotics, the multiple antibiotic resistances of E. faecium, and the increased ability to withstand associated stressors [4, 43].
Genomics of pathogenic enterococci has shown characteristics that distinguish them from commensal isolates [44]. A review by Guzman Prieto et al. (2016) highlights the clonality of clinical enterococci [45]. A comprehensive study of 1644 E. faecium isolates by Arredondo-Alonso et al. (2020) identified the role of plasmids and their subsequent plasmid subpopulations amongst clinical VREfm and non-clinical sources [9]. Comparing isolates from hospitalized patients vs other sources, hospitalized patients carried a larger number of plasmids and plasmids were significantly larger in size. The plasmid population is the largest contributing factor to genome size outside of the core genome, and vast heterogeneity was observed among completed plasmid sequences with respect to plasmid length and number of replication and mobilisation proteins (n = 305). Commensal isolates have smaller genomes, while MDR isolates are promiscuous and have enlarged genomes that include plasmids, phages, insertion sequences (IS), and pathogenicity islands [44]. Enterococci act as an anchoring vector for these mobile genetic elements (MGEs), and up to 25% of enterococcal DNA can be accounted for by acquiring exogenous DNA through these mechanisms. A 2010 study identified that MDR isolates lack functional CRISPR systems [40], which enables MGE uptake. However, the largest study to date reported no difference between number of CRISPR-Cas systems and instead, found a type I restriction modification system (RM) enriched in clade A1 of E. faecium [9]. Separate RM S-subunits were enriched outside this clade suggesting it is the presence of RMs that dictate gene transfer events and drive subspecies separation [9, 46].
Fluctuation analysis of enterococcal genes described a favourable gain in niche-specific genes. Capturing MGEs and acquiring AMR genes in VRE has allowed gene-mediated survival within the hospital, where further MGEs can be acquired ad-hoc [47]. A recent analysis of the core genome of 973 global clade A1 (hospital associated) E. faecium isolates from 31 countries spanning 30 years defined 10 clusters. Low granularity was observed between groups, highlighted by core-genome admixture, which showed substantial ancestry between 78 isolates found at the boundaries, likely due to recombination events [17]. Similarly, a pan-genome analysis identified a significant number of shared genes among plasmids (40.9%), indicating plasmid-driven strain diversification among hospital clones. Low-frequency genes were also observed among plasmids across the pan-plasmidome, suggesting the acquisition of ectopic DNA to the accessory genome. Within the core genome, homologous housekeeping genes with > 5% divergence (adk, atpA and pstS) were observed, but high overall homology indicated clonal expansion of clade A1 [17].
A review by Hendrickx et al. (2013) summarises the role that enterococcal surface proteins play in the pathogenesis of E. faecium [48]. A large set of these proteins are anchored to the cell wall through a LPxTG domain and hence are exposed on the outside of the cell wall. These proteins can represent a pool of surface antigens for therapeutic exploitation that will be discussed later. Several virulence factors exist among enterococci, allowing persistence, evasion and competition among niche co-occupants. Haemolysin (cytolysin), a secreted toxin capable of lysing red and white blood cells, is often encoded in pheromone-responsive plasmids or pathogenicity islands and is associated with increased virulence among VREfs [49, 50]. Of note, the genetic capability to produce cytolysin was found in E. faecium via PCR but was determined to be a silent gene [51].
Gelatinase (gelE), found in > 90% of clinically associated clonal complex 17 (CC17) isolates, and other serine proteinases are responsible for degrading host tissues comprised of collagen to provide nutrients and can affect intestinal epithelial translocation [52, 53]. Gelatinase also modulates the host immune response and activates autolysin, which leads to the fratricidal release of extracellular DNA, a component in biofilm formation [54, 55]. GelE is found in both VREfm and vancomycin-resistant Enterococcus faecalis (VREfs) [56]. Hyaluronidase (hyl) degrades mucopolysaccharides of host connective tissue and extracellular matrix, enabling the spread of the cells and their toxins through host tissue whilst simultaneously providing a disaccharide carbon source. Although hyl genes are present among virulent enterococci, it is not a primary mediator of virulence. Aggregation substance (AS) promotes E. faecalis clumping and facilitates adhesion to eukaryotic cells, such as renal epithelial cells. It also mediates aggregate formation during conjugation and helps in high-frequency plasmid transfer and is not found in E. faecium [57]. Esp and Espfm genes are localised on pathogenicity islands within clinically relevant enterococcal species, where Espfm is a distinct marker for the hospital associated lineage CC17, and plays a role in adherence and biofilm formation among abiotic surfaces [58,59,60]. Microbial surface components recognising adhesive matrix molecules (MSCRAMMs) are essential in the early stages of infection. The cell wall-anchored enterococcal adhesins Ace and Acm are also present among clinically relevant E. faecalis and E. faecium, respectively. Transcriptionally expressed in the presence of urine, serum and collagen, and present as pseudogenes in non-clinical (NC) enterococcal spp., Ace-deleted mutants show reduced virulence for UTIs and endocarditis, highlighting their role in pathogenicity [50,
留言 (0)