
記住我







In the United States, nephrolithiasis (NL) is relatively common, affecting approximately 1 in 11 adults (Scales et al., 2012). A study of 12.7 million children in the United States noted a rate of NL of 54.1 cases per 100,000 person-years in 2016 (Ward et al., 2019). Multiple studies have noted that the incidence of NL and nephrocalcinosis (NC) has increased over the past decade, especially in adolescents and female children (Novak et al., 2009; Dwyer et al., 2012; Ward et al., 2019; Li et al., 2020). There is high morbidity associated with NL and NC, including an increased risk of chronic kidney disease (CKD) and kidney failure (Zhe and Hang, 2017). In addition, the economic burden associated with treatment is high with an annual expenditure of over $10 billion in the United States based on data from 2006 (Economic Impact of Urologic Disease, 2012). In adults, obesity, metabolic syndrome, hypertension, and diabetes have been associated with NL (Taylor et al., 2005; Lieske et al., 2006). The etiology of NL and NC is multifactorial and includes both environmental components and genetic components. Compared to adults, there is a relative paucity of studies in children with NL and NC. This review aims to focus on the genetic component of NL and NC in children.
2 Heritability of nephrolithiasis and nephrocalcinosisTwin studies have illustrated 56% heritability for NL with a significantly higher frequency in female twins compared to male twins (Goldfarb et al., 2005; Goldfarb et al., 2018). Twin studies and large population studies have also shown heritability of serum and urine electrolytes, with that of serum calcium and 24-h urine calcium ranging from 33% to 37% and 44%–52%, respectively (Hunter et al., 2002; Moulin et al., 2017). Heritability of 80% has also been noted for serum 25-hydroxy-vitamin D levels (Wjst et al., 2007).
3 Genetic causes of nephrolithiasis and nephrocalcinosis in childrenCausative gene variants have been detected in 11.5%–31.9% of children with NL and NC using high-throughput multiplex PCR with next-generation exon sequencing (Halbritter et al., 2015; Braun et al., 2016; Gefen et al., 2023). Causative variants have been detected in 29.4% of children with NL and NC using whole exome sequencing of 30 genes associated with monogenic NL and NC and in 32.5% of children with NL using whole exome sequencing of 38 genes associated with monogenic NL in a pediatric Chinese cohort.
Children with NL and NC are genotypically heterogenous, but often phenotypically relatively homogenous, and there are subsequently little data on the predictors of genetic childhood NL and NC. The discordance between phenotype and genotype is well illustrated by a study that showed that 23.9% and 7.3% of children with suspected Dent disease and primary hyperoxaluria (PH), respectively, based on their phenotype were found to have disease causing variants in unrelated genes (Cogal et al., 2021). Regarding identified risk factors for genetic NL in children, a Chinese study found the following: positive family history, consanguinity, younger age at onset, presence of concurrent NC, and CKD (Zhao et al., 2022). In contrast, an American study of children with NL and NC found that the only factor predictive of genetic disease was low serum bicarbonate (Gefen et al., 2023).
Given the high diagnostic yield of genetic testing in this population as well as the genetic heterogeneity, phenotypic homogeneity, and lack of well-established predictors of genetic NL and NC, performing targeted genetic testing broadly for children with NL and NC has been suggested. Genetic testing has become more readily available in recent years with high throughput exon sequencing with whole-exome sequencing or exon panels. Genetic testing may provide clinically meaningful information and affect long-term outcomes. According to multiple studies in children with kidney diseases including NL and NC, genetic testing has the power to alter clinical management and surveillance in >40% of cases (Halbritter et al., 2015; Amlie-Wolf et al., 2021; Jayasinghe et al., 2021).
3.1 Conditions with hypercalciuriaMost genetic diseases associated with NL and NC are secondary to hypercalciuria, including those secondary to hypercalcemia, renal phosphate wasting, renal magnesium wasting, distal renal tubular acidosis (RTA), proximal tubulopathies, mixed or variable tubulopathies, Bartter syndrome, hyperaldosteronism and pseudohyperaldosteronism, hyperparathyroidism and hypoparathyroidism, and other causes. Each of these categories will be expanded on in the following subsections.
3.1.1 Hypercalcemia and hypocalcemiaTable 1 contains a list of genetic causes of primary hypercalcemia and hypocalcemia with hypercalciuria and NL and/or NC. These conditions are due to variants in CASR and CYP24A1 and will be discussed in greater detail below.
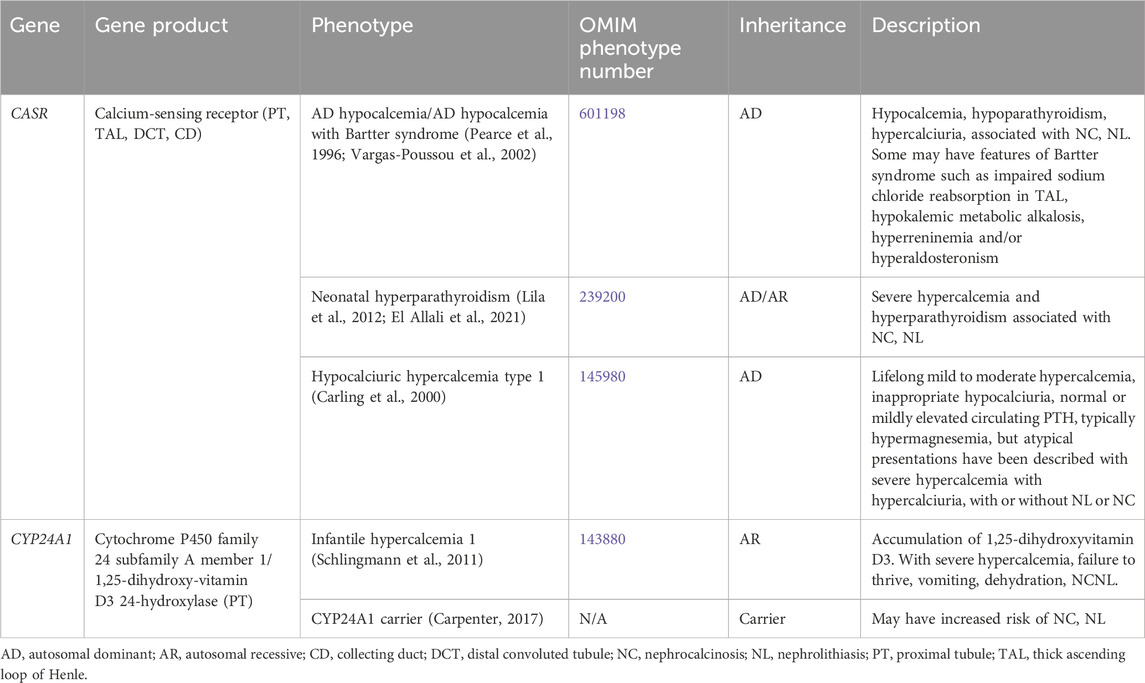
Table 1. Genetic causes of primary hypercalcemia and hypocalcemia with hypercalciuria, nephrolithiasis and/or nephrocalcinosis.
Supplementary Table S1 contains a list of genetic causes of secondary hypercalcemia and hypocalcemia with hypercalciuria and NL and/or NC. Conditions associated with secondary hypercalcemia include Williams-Beuren syndrome (7p11.23, autosomal dominant [AD] inheritance, OMIM phenotype number 194050), Oculoskeletodental syndrome (PIK32CA gene, autosomal recessive [AR] inheritance, OMIM phenotype number 618440) (Tiosano et al., 2019), Blue diaper syndrome (possibly PCSK1 gene, AR versus X-linked recessive [XLR] inheritance, OMIM phenotype number 211000) (Drummond et al., 1964; Distelmaier et al., 2024), Congenital surcease-isomaltase deficiency (SI gene, AR inheritance, OMIM phenotype number 222900) (Starnes and Welsh, 1970; Belmont et al., 2002), and Glucose/galactose malabsorption (SLC5A1 gene, AR inheritance, OMIM phenotype number 606824) (Soylu et al., 2008).
3.1.1.1 CASR geneCASR encodes for the calcium-sensing receptor (CaSR), which is important in renal calcium homeostasis and expressed in the parathyroid gland and the kidney in the thick ascending loop of Henle (TAL) basolateral membrane (Figure 1), the PT, the distal convoluted tubule (DCT), and the CD (Brown and MacLeod, 2001). In the TAL, activation of CaSR by calcium inhibits NaCl reabsorption by inhibiting NKCC2 and ROMK (Gamba and Friedman, 2009). By inhibiting ROMK, the tubular lumen positive voltage is diminished, reducing the driving force for paracellular cation (including calcium) reabsorption (Gamba and Friedman, 2009).
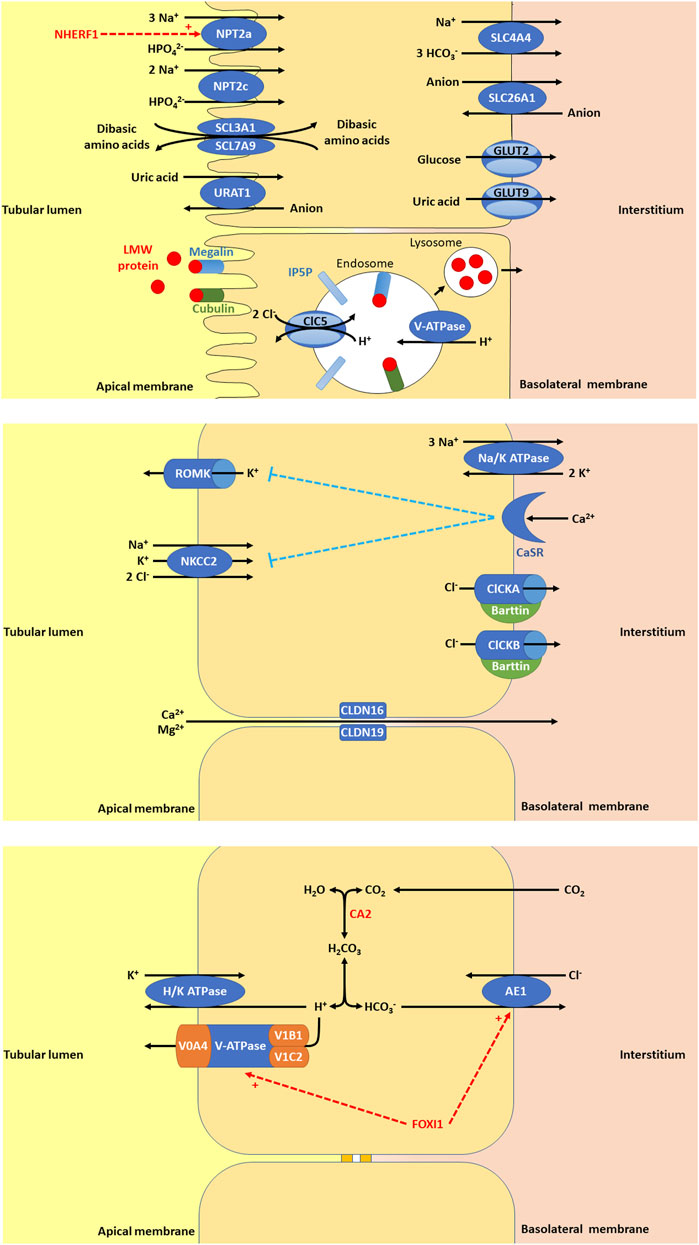
Figure 1. (A) In the proximal tubule (PT), there are a variety of apical membrane transporters, basolateral membrane transporters, and endosome transporters proteins that play important roles in PT function. Apical membrane transporters shown are sodium-dependent phosphate transport protein 2a (NPT2a, encoded by SLC34A1, whose activity is promoted by NHERF1, sodium/hydrogen exchange regulatory factor 1, encoded by SLC9A3R1), sodium-dependent phosphate transport protein 2c (NPT2c, encoded by SLC34A3), SLC3A1/SLC7A9 (encoded by SLC3A1 and SLC7A9), and urate-anion transporter (URAT1, encoded by SLC22A12). The basolateral membrane transporters shown are glucose transporter 2 (GLUT2, encoded for by SLC2A2), facilitated glucose transporter 9 (GLUT9)/voltage-driven urate transporter (encoded for by SLC2A9), solute carrier family 4 member 4 (SLC4A4, a sodium bicarbonate transporter encoded by SLC4A4), and solute carrier family 26 member 1 (SLC26A1, an electroneutral anion exchanger encoded by SLC26A1). Low molecular weight (LMW) protein in the tubular lumen binds megalin and cubulin and are endocytosed into endosomes, thought to be facilitated by inositol polyphosphate-5-phosphatase (IP5P), ended by OCRL, which is expressed in the glomerulus, the PT, the thick ascending loop of Henle (TAL), the distal convoluted tubule (DCT) and the collecting duct (CD). Endosome acidification is then mediated by V-ATPase and the chloride voltage-gated channel 5 (ClC5, encoded by CLCN5, expressed in the PT, alpha-intercalated cell (alpha-IC) and the TAL. Lysosomes form to allow the absorption of LMW protein and recycling of megalin and cubulin to the apical membrane. (B) In the TAL, there are a variety of apical membrane transporters, tight junction proteins, and basolateral membrane transporters, receptors, and proteins that play important roles in tubular function. Apical membrane transporters shown are kidney-specific Na-K-Cl symporter (NKCC2) and renal out-medullar potassium channel (ROMK). NKCC2 (encoded by SLC12A1) that is responsible for sodium, potassium, and chloride reabsorption from the tubular lumen. ROMK (encoded by KCNJ1) is responsible for excretion of potassium. The tight junction proteins shown are claudin 16 (CLDN16, encoded by CLDN16) and claudin 19 (CLDN19, encoded by CLDN19). CLDN16 and CLDN19 are necessary for paracellular reabsorption of calcium and magnesium. The basolateral membrane transporters and receptors shown include Na/K ATPase, chloride voltage-gated channel kidney A (ClCKA), chloride voltage-gated channel kidney B (ClCKB), Barttin, and calcium sensing receptor (CaSR). Na/K ATPase is responsible for generating the gradient for sodium entry across apical membranes via NKCC2 and subsequent exit of chloride across the basolateral membrane via ClCKA, ClCKB, and Barttin. ClCKA is encoded for by CLCNKA and ClCKB is encoded for by CLCNKB. These chloride channels allow chloride to exit the cell via the basolateral membrane. Barttin or chloride voltage-gated channel kidney (ClCK)-type accessory subunit beta, encoded for by BSND. ClCKA and ClCKB channels depend on the presence of the Barttin subunit for chloride transport. CaSR (encoded for by CASR), is expressed in the parathyroid gland and the kidney in the TAL, the PT, the DCT, and the CD. In the TAL, activation of CaSR by calcium inhibits NaCl reabsorption by inhibiting NKCC2 and ROMK. By inhibiting ROMK, the tubular lumen positive voltage is diminished, reducing the driving force for paracellular cation (including calcium) reabsorption. (C) In the alpha-IC of the CD, there are few key, apical membrane transporters, and basolateral membrane transporters that play important roles in tubular function. Carbonic anhydrase 2 (CA2) is an enzyme encoded by CA2 that is expressed in the CD (including the alpha-IC) and PT. CA2 catalyzes the combination of carbon dioxide and water to form carbonic acid (H2CO3), which then dissociates to protons (H+) and bicarbonate (HCO3−). The protons produced are excreted into the urine by exiting the cell through the apical membrane via H/K ATPase and V-ATPase, a proton transporter with V0 subunit A4 (V0A4, encoded by ATP6V0A4), V0 subunit B1 (V1B1, encoded by ATP6V1A1), and V1 subunit C2 (V1C2, encoded by ATP6V1C2). The bicarbonate produced is reabsorbed into the interstitium by exiting the cell thought the basolateral membrane via anion exchange protein 1 (AE1), a basolateral chloride bicarbonate counter transporter encoded by SLC4A1. Forkhead box I1 (FOXI1) encoded for by FOXI1 is a transcription factor that regulates the function of AE1 and V-ATPase.
AD hypocalcemia/AD hypocalcemia with Bartter syndrome (OMIM phenotype number 601198) is a condition resulting from activating variants in CASR, which increase the sensitivity of CaSR to extracellular calcium (Brown and MacLeod, 2001). Activation of CaSR in the parathyroid gland leads to inhibition of parathyroid hormone (PTH) release and subsequent hypoparathyroidism (Brown and MacLeod, 2001). Reduced PTH release leads to decreased calcium and phosphorus resorption in the bone (Brown and MacLeod, 2001). In the kidney, reduced PTH leads to decreased TAL and DCT reabsorption of calcium, increased reabsorption of phosphorus, and decreased production of 1,25-dihydroxyvitamin D3, leading to decreased absorption of calcium in the intestine (Brown and MacLeod, 2001). All of the above culminates in hypocalcemia, hypercalciuria, NC, and NL (Pearce et al., 1996). Some may have features of Bartter syndrome such as impaired sodium chloride reabsorption in the TAL, hypokalemic metabolic alkalosis, hyperreninemia and/or hyperaldosteronism (Vargas-Poussou et al., 2002). Treatment with minimal doses of calcium and vitamin D administration may be given to alleviate symptoms, but excess can exacerbate hypercalciuria, NC, and lead to kidney impairment (Pearce et al., 1996).
Neonatal hyperparathyroidism (OMIM phenotype number 239200) is an AD/AR condition resulting from inactivating variants in CASR that decrease the sensitivity of CaSR to extracellular calcium (Brown and MacLeod, 2001). Inactivation of CaSR in the parathyroid gland leads to stimulation of PTH release and subsequent hyperparathyroidism (Lila et al., 2012; El Allali et al., 2021). Increased PTH release leads to increased calcium and phosphorus resorption in the bone (Lila et al., 2012; El Allali et al., 2021). In the kidney, increased PTH leads to increased tubular reabsorption of calcium, decreased reabsorption of phosphorus, and increased production of 1,25-dihydroxyvitamin D3, leading to increased absorption of calcium in the intestine (Lila et al., 2012; El Allali et al., 2021). This culminates in severe hypercalcemia, NC, and NL (Lila et al., 2012; El Allali et al., 2021). Calcimimetic medications may be given treat this condition by increasing sensitivity of CaSR to extracellular calcium (El Allali et al., 2021).
Hypocalciuric hypercalcemia type 1 (OMIM phenotype number 145980) is an AD condition resulting from an inactivating variant in CASR, resulting in lifelong mild to moderate hypercalcemia, inappropriate hypocalciuria, and normal PTH to mild hyperparathyroidism (Carling et al., 2000). This condition may also have atypical presentations with severe hypercalcemia and hypercalciuria with or without NL or NC (Carling et al., 2000).
3.1.1.2 CYP24A1 geneCYP24A1 encodes for cytochrome P450 family 24 subfamily A member 1 or 1,25-dihydroxyvitamin D3 24-hydroxylase, the enzyme primarily responsible for the catabolism of 1,25-dihydroxy- and 25-hydroxy-vitamin D, which is expressed in the PT (Schlingmann et al., 2011). Infantile hypercalcemia 1 (OMIM phenotype number 143880) is an AR condition resulting from an inactivating variant in CYP24A1, resulting in accumulation of 1,25-dihydroxyvitamin D3 (Schlingmann et al., 2011). This accumulation leads to severe hypercalcemia, failure to thrive, vomiting, dehydration, NC, and NL (Schlingmann et al., 2011). Data suggests that carriers (heterozygotes) of inactivating variants in CYP24A1 may have increased risk of NC and NL (Carpenter, 2017).
For treatment of Infantile hypercalcemia 1, generous hydration and a diet low in vitamin D and calcium are suggested (Schlingmann et al., 2011). CYP3A4 inducers (rifampin) or CYP27B1 modulators (fluconazole/ketoconazole/itraconazole) may help reduce 1,25-dihydroxyvitamin D3 levels and improve hypercalcemia and hypercalciuria (Sayers et al., 2015; Hawkes et al., 2017). Low doses of these medications are suggested, but the long-term safety is uncertain.
3.1.2 Renal phosphate wasting with hypercalciuriaGenetic causes of renal phosphate wasting with hypercalciuria and NL and/or NC are due to variants in SCL34A1, SLC34A3, and SLC9A3R1 (Table 2). These individuals typically require treatment with phosphate supplementation (Tieder et al., 1992).
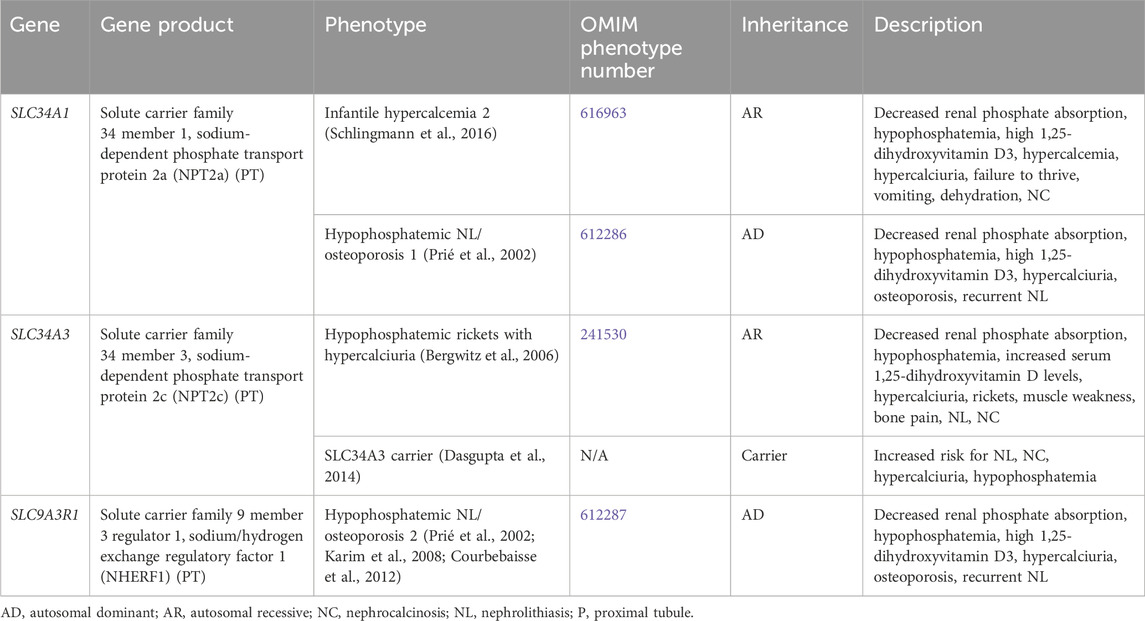
Table 2. Genetic causes of renal phosphate wasting with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.2.1 SLC34A1 geneSLC34A1 encodes for the solute carrier family 34 member 1, the sodium-dependent phosphate transport protein 2a (NPT2a), which responsible for the majority of renal phosphate transport and is primarily expressed in the PT (Figure 1) (Schlingmann et al., 2016). Fanconi renotubular syndrome 2 (OMIM phenotype number 613388) is one of the conditions caused by homozygous variants in SLC34A1 but will not be discussed in this review as this condition has not been associated with NL or NC.
Infantile hypercalcemia 2 (OMIM phenotype number 616963) is an AR condition due to inactivating variants in SLC34A1, resulting in loss of NPT2a activity and subsequent renal phosphorus wasting with hypophosphatemia (Schlingmann et al., 2016). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalcemia, hypercalciuria, failure to thrive, vomiting, dehydration, and NC (Schlingmann et al., 2016). In addition to phosphate supplementation, generous hydration and a diet low in vitamin D and calcium are also suggested (Schlingmann et al., 2016). CYP3A4 inducers (rifampin) or CYP27B1 modulators (fluconazole/ketoconazole/itraconazole) may help reduce 1,25-dihydroxyvitamin D3 levels and improve hypercalcemia and hypercalciuria (Sayers et al., 2015; Hawkes et al., 2017). Low doses of these medications are suggested, but the long-term safety is uncertain.
Hypophosphatemic NL/osteoporosis 1 (OMIM phenotype number 612286) is an AD condition due to an inactivating variant in SLC34A1, resulting in loss of NPT2a activity and subsequent renal phosphorus wasting with hypophosphatemia (Prié et al., 2002). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalciuria, osteoporosis, and recurrent NL (Prié et al., 2002).
3.1.2.2 SLC34A3 geneSLC34A3 encodes for the solute carrier family 34 member 3, the sodium-dependent phosphate transport protein 2c (NPT2c) which is primarily expressed in the PT (Figure 1) (Bergwitz et al., 2006). Hypophosphatemic rickets with hypercalciuria (OMIM phenotype number 241530) is an AR condition due to inactivating variants in SLC34A3, resulting in loss of NPT2c activity and subsequent renal phosphorus wasting with hypophosphatemia (Bergwitz et al., 2006). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalcemia, hypercalciuria, rickets, muscle weakness, bone pain, NL, and NC (Bergwitz et al., 2006). Data suggests that carriers (heterozygotes) of inactivating variants in SLC34A3 may have increased risk of NL, NC, hypercalciuria, and hypophosphatemia (Dasgupta et al., 2014).
3.1.2.3 SLC9A3R1 geneSLC9A3R1 encodes for the solute carrier family 9 member 3 regulator 1/sodium/hydrogen exchange regulatory factor 1 (NHERF1), which is primarily expressed in the PT (Figure 1) (Karim et al., 2008; Courbebaisse et al., 2012). Hypophosphatemic NL/osteoporosis 2 (OMIM phenotype number 612287) is an AD condition due to an inactivating variant in SLC9A3R1, resulting in loss of NHERF1 activity with subsequent decrease in NPT2a expression, resulting in renal phosphorus wasting with hypophosphatemia (Courbebaisse et al., 2012). This subsequently results in increased production of 1,25-dihydroxyvitmain D3 with hypercalciuria, osteoporosis, and recurrent NL (Prié et al., 2002).
3.1.3 Renal magnesium wasting with hypercalciuriaGenetic causes of renal magnesium wasting with hypercalciuria and NL and/or NC are related to, CLDN16, CLDN19, and RRAGD, and are shown in Table 3. The conditions related to variants in these genes are discussed further below.
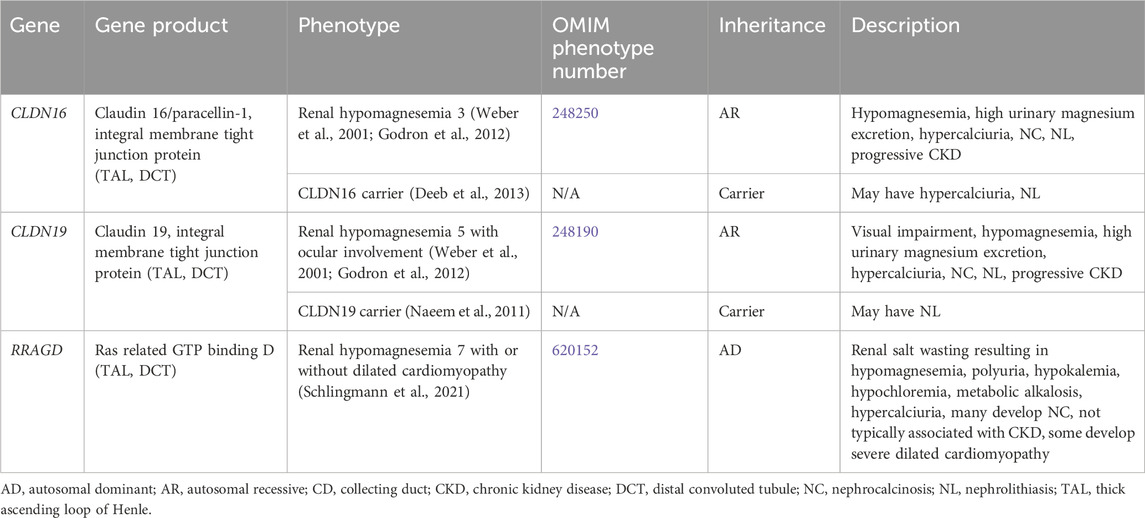
Table 3. Genetic causes of renal magnesium wasting with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.3.1 CLDN16 geneCLDN16 encodes for Claudin 16 or paracellin-1, which is an integral membrane tight junction protein necessary for paracellular reabsorption of calcium and magnesium (Godron et al., 2012). CLDN16 is expressed in the TAL and DCT (Figure 1). Renal hypomagnesemia 3 (OMIM phenotype number 248250) is an AR condition due to inactivating variants in CLDN16, resulting reduced reabsorption of calcium (hypercalciuria)and magnesium (hypermagnesuria) with hypomagnesemia, NC, NL, and progressive CKD (Godron et al., 2012). Data suggests that carriers (heterozygotes) of inactivating variants in CLDN16 may have hypercalciuria and NL (Deeb et al., 2013).
Medical management of Renal hypomagnesemia 3 typically involves high-dose magnesium supplementation to replace renal losses, although this has not been shown to slow progression of CKD (Weber et al., 2001; Godron et al., 2012). Thiazide diuretics and indomethacin have been used in this condition but have not been shown to be successful in reducing hypercalciuria or hypermagnesuria or preventing progression of CKD (Weber et al., 2001; Godron et al., 2012). Definitive treatment can be achieved with kidney transplantation (Weber et al., 2001).
3.1.3.2 CLDN19 geneCLDN19 encodes for Claudin 19, which is an integral membrane tight junction protein necessary for paracellular reabsorption of calcium and magnesium (Godron et al., 2012). CLDN19 is expressed in the TAL and DCT (Figure 1). Renal hypomagnesemia 5 with ocular involvement (OMIM phenotype number 248190) is an AR condition due to inactivating variants in CLDN19, resulting reduced reabsorption of calcium (hypercalciuria) and magnesium (hypermagnesuria)with hypomagnesemia, visual impairment, NC, NL, and progressive CKD (Godron et al., 2012). Data suggests that carriers (heterozygotes) of inactivating variants in CLDN16 may have NL (Naeem et al., 2011). Medical management of Renal hypomagnesemia 5 with ocular involvement and its efficacy are typically the same as that for Renal hypomagnesemia 3 caused by variants in CLDN16 (Weber et al., 2001; Godron et al., 2012).
3.1.3.3 RRAGD geneRRAGD encodes for Ras related GTP binding D, which is expressed in the TAL and DCT (Schlingmann et al., 2021). Renal hypomagnesemia 7 with or without dilated cardiomyopathy (OMIM phenotype number 620152) is an AD condition due to an inactivating variant in RRAGD, resulting in activation of mTOR signaling, which leads to renal salt wasting resulting in hypomagnesemia, polyuria, hypokalemia, hypochloremia, metabolic alkalosis, hypercalciuria, and NC (Schlingmann et al., 2021). This condition is not typically associated with CKD, and some individuals develop severe dilated cardiomyopathy (Schlingmann et al., 2021). Optimal treatment strategy is unclear.
3.1.4 Distal renal tubular acidosisDistal RTA results from the impaired ability to acidify the urine, which in turn leads to hyperchloremic metabolic acidosis (Karet, 2002). Distal RTA is associated and hypocitraturia with alkaline urine due to upregulation of citrate reabsorption in the PT, hypercalciuria and resultant NC and/or NL, and may be associated with hypokalemia (Karet, 2002; Lopez-Garcia et al., 2019). Treatment for distal RTA primarily consists of alkali therapy to correct metabolic acidosis, with potassium-containing medications such as potassium citrate being the preferred choice (Mohebbi and Wagner, 2018). Thiazide diuretics may be considered with severe hypercalciuria but may be complicated by polyuria and hypokalemia (Mohebbi and Wagner, 2018). Many individuals with chronic untreated or severe metabolic acidosis develop rickets or osteomalacia, although it appears as though most adults achieve normal adult height, especially those with better control of acidosis (Karet, 2002; Lopez-Garcia et al., 2019). CKD stages 2–4 have been reported in >80% of adults and in >30% of children with distal RTA with ESKD rarely occurring (Lopez-Garcia et al., 2019). In individuals with better control of acidosis, CKD incidence is lower (Lopez-Garcia et al., 2019). Table 4 shows genetic causes of distal RTA with hypercalciuria and NL and/or NC.
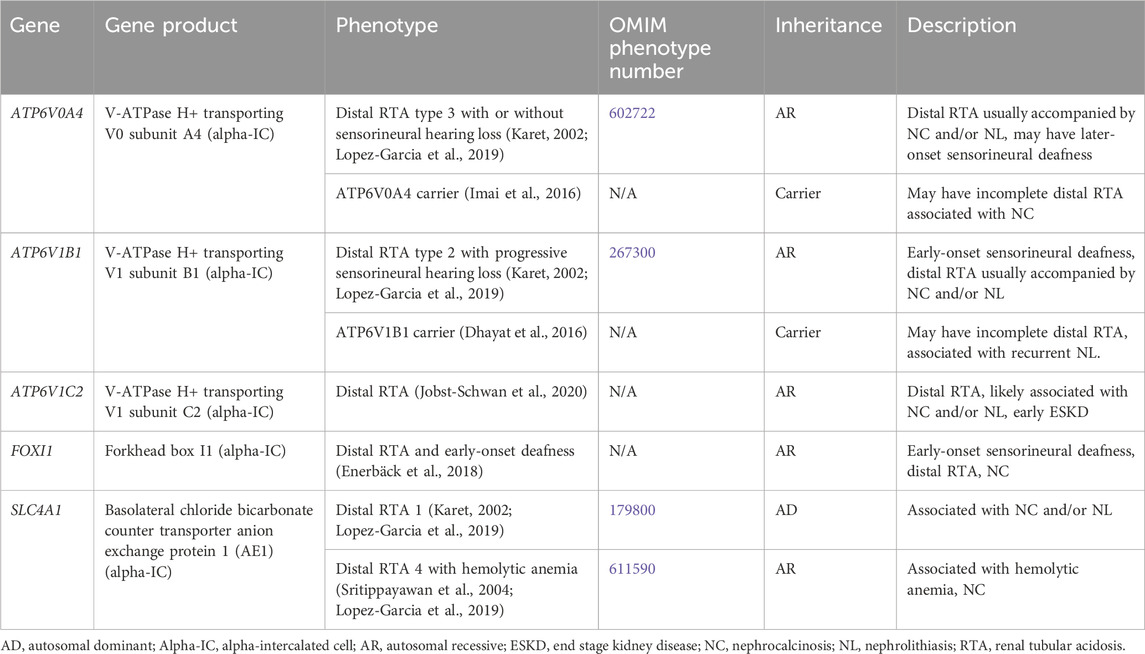
Table 4. Genetic causes of distal renal tubular acidosis with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.4.1 ATP6V0A4 geneATP6V0A4 encodes for V-ATPase H+ transporting V0 subunit A4, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA type 3 with or without sensorineural hearing loss (OMIM phenotype number 602722) is an AR condition due to inactivating variants in ATP6V0A4, resulting in decreased urinary proton excretion, which leads to a distal RTA usually accompanied by NC and/or NL as well later-onset sensorineural deafness in >30% (Karet, 2002; Lopez-Garcia et al., 2019). Data suggests that carriers (heterozygotes) of inactivating variants in ATP6V0A4 may have an incomplete distal RTA associated with NC (Imai et al., 2016).
3.1.4.2 ATP6V1B1 geneATP6V1B1 encodes for V-ATPase H+ transporting V1 subunit B1, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA type 2 with progressive sensorineural hearing loss (OMIM phenotype number 267300) is an AR condition due to inactivating variants in ATP6V1B1, resulting in decreased urinary proton excretion, which leads to a distal RTA usually accompanied by NC and/or NL as well early-onset sensorineural deafness in most (Lopez-Garcia et al., 2019). Data suggests that carriers (heterozygotes) of inactivating variants in ATP6VAB1 may have an incomplete distal RTA associated with recurrent NL (Dhayat et al., 2016).
3.1.4.3 ATP6V1C2 geneATP6V1C2 encodes for V-ATPase H+ transporting V1 subunit C2, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Jobst-Schwan et al., 2020). ATP6V1C2-associated distal RTA is an AR condition due to inactivating variants in ATP6V1C2, resulting in decreased urinary proton excretion, which leads to a distal RTA that is likely associated with NC and/or NL and with early ESKD (Jobst-Schwan et al., 2020).
3.1.4.4 SLC4A1 geneSLC4A1 encodes for the basolateral chloride bicarbonate counter transporter anion exchange protein 1 (AE1), which reabsorbs bicarbonate from the urine into the circulation and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA 1 (OMIM phenotype number 179800) is an AD condition due to an inactivating variant in SLC4A1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA usually accompanied by NC and NL (Karet, 2002; Lopez-Garcia et al., 2019). Distal RTA 4 with hemolytic anemia (OMIM phenotype number 611590) is an AR condition due to inactivating variants in SLC4A1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA with hemolytic anemia and NC. (Sritippayawan et al., 2004; Lopez-Garcia et al., 2019).
3.1.4.5 FOXI1 geneFOXI1 encodes for Forkhead box I1, which is a transcription factor that regulates the function of AE1, AE4, and V-ATPase subunits, and is expressed in the alpha-IC of the CD (Figure 1) (Enerbäck et al., 2018). Distal RTA and early-onset deafness is an AR condition due to inactivating variants in FOXI1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA with early-onset sensorineural deafness and NC (Enerbäck et al., 2018).
3.1.5 Proximal tubulopathyProximal tubulopathy consists of dysfunction of the PT, which can lead to any combination of low molecular weight (LMW) proteinuria, aminoaciduria, glucosuria, urine bicarbonate wasting and RTA, hypercalciuria, hyperphosphaturia, high urinary potassium excretion, and uricosuria. NC and NL can occur with proximal tubulopathies, but less commonly so than with distal RTA. Another name for proximal tubulopathy is Fanconi renotubular syndrome. Treatment generally consists of supplementation to treat urinary solute losses.
Supplementary Table S2 contains a list of genetic causes of secondary proximal tubulopathy with hypercalciuria and NL and/or NC. Conditions that results in a secondary proximal tubulopathy include Hereditary fructose intolerance (ALDOB gene, AR inheritance (Higgins and Varney, 1966; Mass et al., 1966), OMIM phenotype number 229600), Wilson disease (ATP7B gene, AR inheritance, OMIM phenotype number 277900) (Di Stefano et al., 2012), Nephropathic cystinosis (CTNS gene, AR inheritance, OMIM phenotype number 219800) (Theodoropoulos et al., 1995), Tyrosinemia type 1 (FAH gene, AR inheritance, OMIM phenotype number 276700) (Forget et al., 1999), Congenital lactase deficiency (LCT gene, AR inheritance, OMIM phenotype number 223000) (Saarela et al., 1995), Mitochondrial DNA depletion syndrome 8A (encephalomyopathic type with renal tubulopathy) (RRM2B gene, AR inheritance, OMIM phenotype number 612075) (Finsterer and Scorza, 2017), Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (TRNT1 gene, AR inheritance, OMIM phenotype number 616084) (Wiseman et al., 2013), Arthrogryposis, renal dysfunction, and cholestasis 1 (VPS33B gene, AR inheritance, OMIM phenotype number 208085) (Holme et al., 2013), and Arthrogryposis, renal dysfunction, and cholestasis 2 (VIPAS39 gene, AR inheritance, OMIM phenotype number 613404) (Holme et al., 2013).
Primary inherited proximal tubulopathies associated with NC and/or NL are shown in Table 5 and will be discussed in greater detail below. Inactivating variants in EHHADH (encodes for enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase) cause AD Fanconi renotubular syndrome 3 (OMIM phenotype number 615605), is associated with rickets, impaired growth, glucosuria, generalized aminoaciduria, phosphaturia, metabolic acidosis, LMW proteinuria, and hypercalciuria will not be discussed as there is no known association with NC or NL (Tolaymat et al., 1992; Klootwijk et al., 2014).
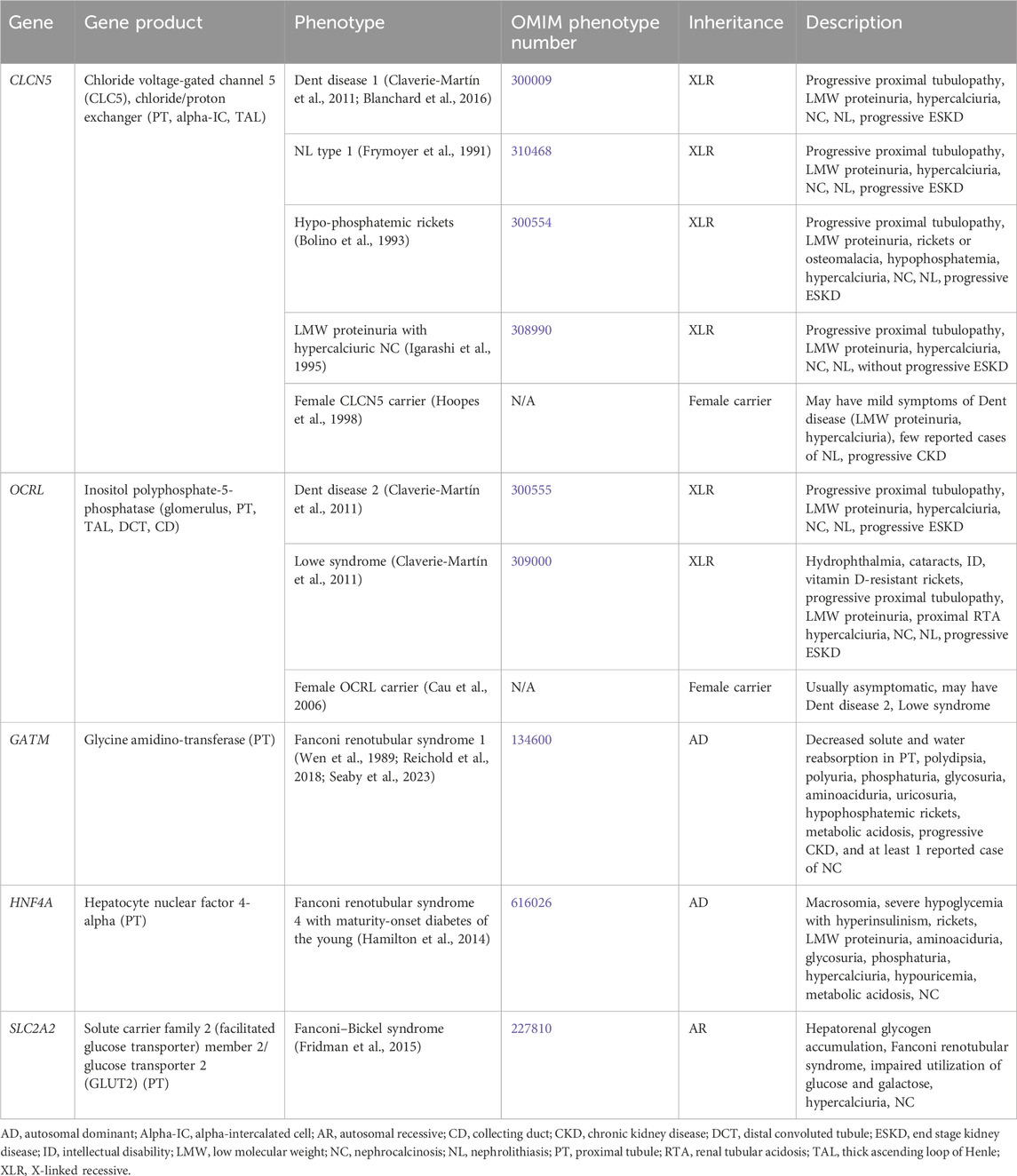
Table 5. Genetic causes of primary proximal tubulopathy with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.5.1 CLCN5 geneCLCN5 encodes for chloride voltage-gated channel 5 (CLC5), a chloride/proton exchanger, and is expressed in the PT (Figure 1), alpha-IC, and TAL (Mansour-Hendili et al., 2015). Inactivating variants in CLCN5 appear to cause abnormal reabsorption of LMW proteins by disrupting PT vesicle acidification, impacting lysosome function and LMW protein processing (Nielsen et al., 2016). Inactivating variants in CLCN5 are associated with a spectrum of conditions (Claverie-Martín et al., 2011).
Dent disease 1 (OMIM phenotype number 300009) is an XLR condition resulting from inactivating variants in CLCN5. Hallmark features are progressive proximal renal tubulopathy with LMW proteinuria, hypercalciuria, NC, NL, and progressive ESKD (Claverie-Martín et al., 2011). Median age at ESKD onset has been reported as 40 years of age (Blanchard et al., 2016). Treatment is aimed at treating serum electrolyte disturbances (such as treating hypophosphatemia with phosphorus supplementation) and minimizing hypercalciuria, primarily with hydration and administration of thiazide diuretics. However, the administration thiazide diuretics to reduce hypercalciuria is controversial as there is no definite correlation between hypercalciuria and ESKD, and significant side effects have been reported including dehydration and hypokalemia (Blanchard et al., 2008). Kidney transplantation has been successful in these patients without recurrence of NL or NC (Scheinman, 1998). Due to the risk of worsening hypercalciuria with vitamin D supplementation, it should be reserved for patients who require it for treatment of rickets.
NL type 1 (OMIM phenotype number 310468) is an XLR condition resulting from inactivating variants in CLCN5. Hypophosphatemic rickets (OMIM phenotype number 300554) is an XLR condition resulting from inactivating variants in CLCN5. It is characterized by progressive proximal renal tubulopathy with LMW proteinuria, rickets or osteomalacia, hypophosphatemia, hypercalciuria, NC, NL, and progressive ESKD (Bolino et al., 1993). LMW proteinuria with hypercalciuric NC (OMIM phenotype number 308990) is an XLR condition resulting from inactivating variants in CLCN5. The presentation of LMW proteinuria with hypercalciuric NC resembles that of the conditions mentioned above, except that these individuals do not experience progressive ESKD (Igarashi et al., 1995). Although these three conditions have different OMIM phenotype numbers, it is unclear if they are truly separate conditions from Dent disease 1 as the spectrum of presenting symptoms are the same (Frymoyer et al., 1991). Data suggests that female carriers of inactivating variants in CLCN5 may have mild symptoms of Dent disease such as low-molecular-weight proteinuria, and hypercalciuria with few reported cases of NL and kidney insufficiency (Hoopes et al., 1998).
3.1.5.2 OCRL geneOCRL encodes for inositol polyphosphate-5-phosphatase, which is thought to be important in endocytosis and cellular trafficking and is expressed throughout the body, including in the glomerulus, PT (Figure 1), TAL, DCT, and CD (Claverie-Martín et al., 2011). Inactivating variants in OCRL appear to cause abnormal protein trafficking required for PT solute reabsorption (Ooms et al., 2009). Inactivating variants in OCRL are associated with a spectrum of conditions (Claverie-Martín et al., 2011).
Dent disease 2 (OMIM phenotype number 300555) is an XLR condition resulting from inactivating variants in OCRL. Clinical features are shared with that of Dent disease 1 caused by variants in CLCN5 with progressive proximal renal tubulopathy with LMW proteinuria, hypercalciuria, NC, NL, and progressive ESKD (Claverie-Martín et al., 2011). Lowe syndrome (OMIM phenotype number 309000) is an XLR condition resulting from inactivating variants in OCRL. The presentation resembles that of Dent disease 2 but also involves other systemic symptoms of hydrophthalmia, cataracts, severely impaired intellectual development, vitamin D-resistant rickets, and proximal RTA (Claverie-Martín et al., 2011). Data suggests that female carriers of inactivating variants in ORCL are usually asymptomatic, but may have either Dent disease 2 or Lowe syndrome (Cau et al., 2006). Treatment of Dent disease 2 and Lowe syndrome is the same as for Dent disease 1, including treatment of electrolyte derangements such as acidosis.
3.1.5.3 GATM geneGATM encodes for glycine amidinotransferase, which is expressed in the PT mitochondria (Reichold et al., 2018). An inactivating variant in GATM leads to Fanconi renotubular syndrome 1 (OMIM phenotype number 134600), an AD condition associated with decreased solute and water reabsorption in the PT with polydipsia, polyuria, phosphaturia, glycosuria, aminoaciduria, uricosuria, hypophosphatemic rickets, metabolic acidosis, progressive kidney insufficiency, and NC in 1 definitive case, and at least 1 case of AD Fanconi syndrome suspected to be secondary a variant in GATM, although genetic testing was not performed (Wen et al., 1989; Reichold et al., 2018; Seaby et al., 2023).
3.1.5.4 HNF4A geneHNF4A encodes for hepatocyte nuclear factor 4-alpha, which in the kidney is expressed in the PT. An inactivating variant in HNF4A leads to Fanconi renotubular syndrome 4 with maturity-onset diabetes of the young (OMIM phenotype number 616026), an AD condition associated with macrosomia, severe hypoglycemia with hyperinsulinism, rickets, LMW proteinuria, aminoaciduria, glycosuria, phosphaturia, hypercalciuria, hypouricemia, metabolic acidosis, and NC (Hamilton et al., 2014).
3.1.5.5 SLC2A2 geneSLC2A2 encodes for solute carrier family 2 (facilitated glucose transporter) member 2 or glucose transporter 2 (GLUT2), which mediates monosaccharide bidirectional transport in the liver, pancreas, intestines, and the renal PT (Figure 1) (Fridman et al., 2015). Inactivating variants in SLC2A2 lead to Fanconi–Bickel syndrome (OMIM phenotype number 227810), an AR condition associated with Fanconi renotubular syndrome, hepatorenal glycogen accumulation, impaired utilization of glucose and galactose, hypercalciuria, and NC (Fridman et al., 2015).
3.1.6 Mixed or variable tubulopathyGenetic causes of mixed or variable tubulopathy with hypercalciuria and NL and/or NC are shown in Table 6. One such condition, Kearns-Sayre syndrome, is associated with various mitochondrial DNA (mtDNA) deletions and a variety of tubulopathies (AR inheritance, OMIM phenotype number 530000) (Finsterer and Scorza, 2017). Treatment for distal RTA primarily consists of alkali therapy to correct metabolic acidosis, and treatment of proximal tubulopathy primarily consists of replacement or urinary solute losses (Mohebbi and Wagner, 2018).
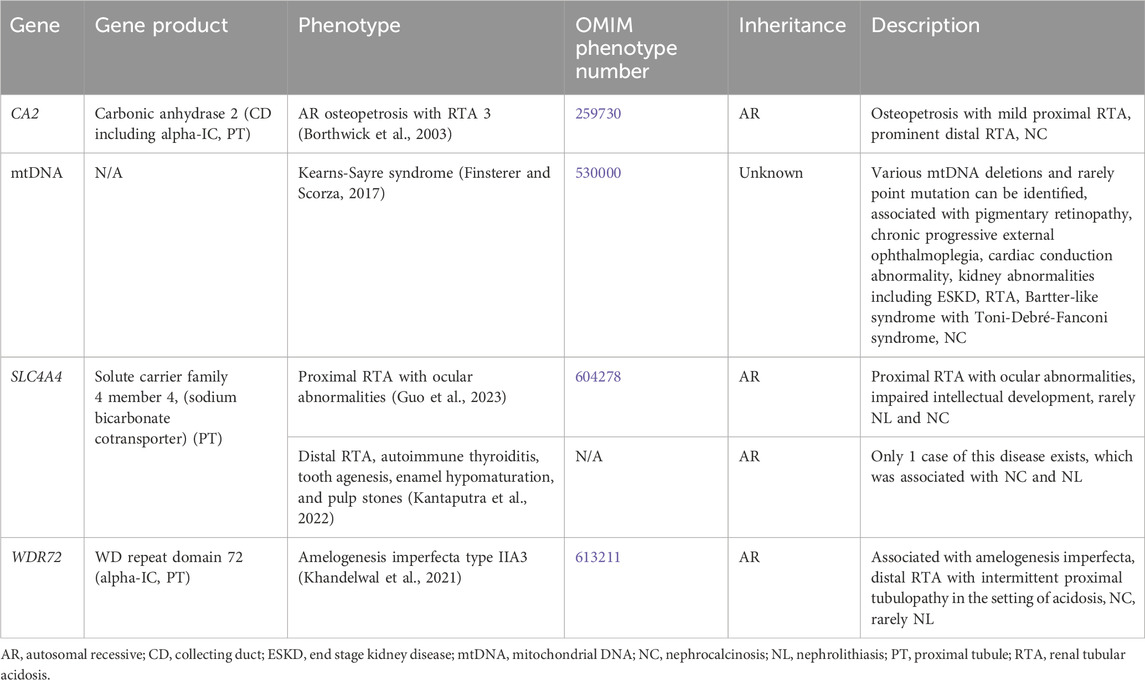
Table 6. Genetic causes of mixed or variable tubulopathy with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.6.1 CA2 geneCA2 encodes for carbonic anhydrase 2 (CA2), which is expressed in the CD (including the alpha-IC) and PT (Figure 1). CA2 catalyzes the combination of carbon dioxide and water to form carbonic acid, which then dissociates to protons and bicarbonate (Whyte, 1993). Inactivating variants in CA2 lead to decreased proton and bicarbonate production in osteoclasts and the renal tubules and is associated AR Osteopetrosis with RTA 3 (OMIM phenotype number 259730). This condition is associated with both a proximal RTA and a distal RTA (Borthwick et al., 2003).
3.1.6.2 SLC4A4 geneSLC4A4 encodes for solute carrier family 4 member 4, a sodium bicarbonate cotransporter that is important in the reabsorption of bicarbonate in the PT (Figure 1) (Guo et al., 2023). Inactivating variants in this gene are associated with two different conditions. Proximal RTA with ocular abnormalities (OMIM phenotype number 604278) is an AR condition associated with impaired intellectual development, and rarely NL and NC (Guo et al., 2023). Distal RTA, autoimmune thyroiditis, tooth agenesis, enamel hypomaturation, and pulp stones is an AR condition with only 1 case reported with the only case being associated with NC and NL (Kantaputra et al., 2022).
3.1.6.3 WDR72 geneWDR72 encodes for WD repeat domain 72, implicated in the trafficking of V-ATPases and thought to play a role in sustained intracellular CaSR signaling through clathrin-mediated endocytosis (Khandelwal et al., 2021). WDR72 is expressed in the alpha-IC in the CD and PT (Khandelwal et al., 2021). Inactivating variants lead to the AR condition Amelogenesis imperfecta type IIA3 (OMIM phenotype number 613211) associated with a distal RTA and intermittent proximal tubulopathy in the setting of acidosis (Khandelwal et al., 2021). This condition is associated with NC and rarely NL.
3.1.7 Bartter syndromeBartter syndrome consists of a group of channelopathies that affect transporter proteins primarily in the TAL involved in sodium chloride reabsorption, which lead to urinary sodium losses, metabolic alkalosis, hypokalemia, hyperaldosteronism and/or hyperreninemia, and hypercalciuria due to loss of paracellular calcium reabsorption (Besouw et al., 2020). There are multiple forms of Bartter syndrome, all of which have been associated with NC (Table 7).
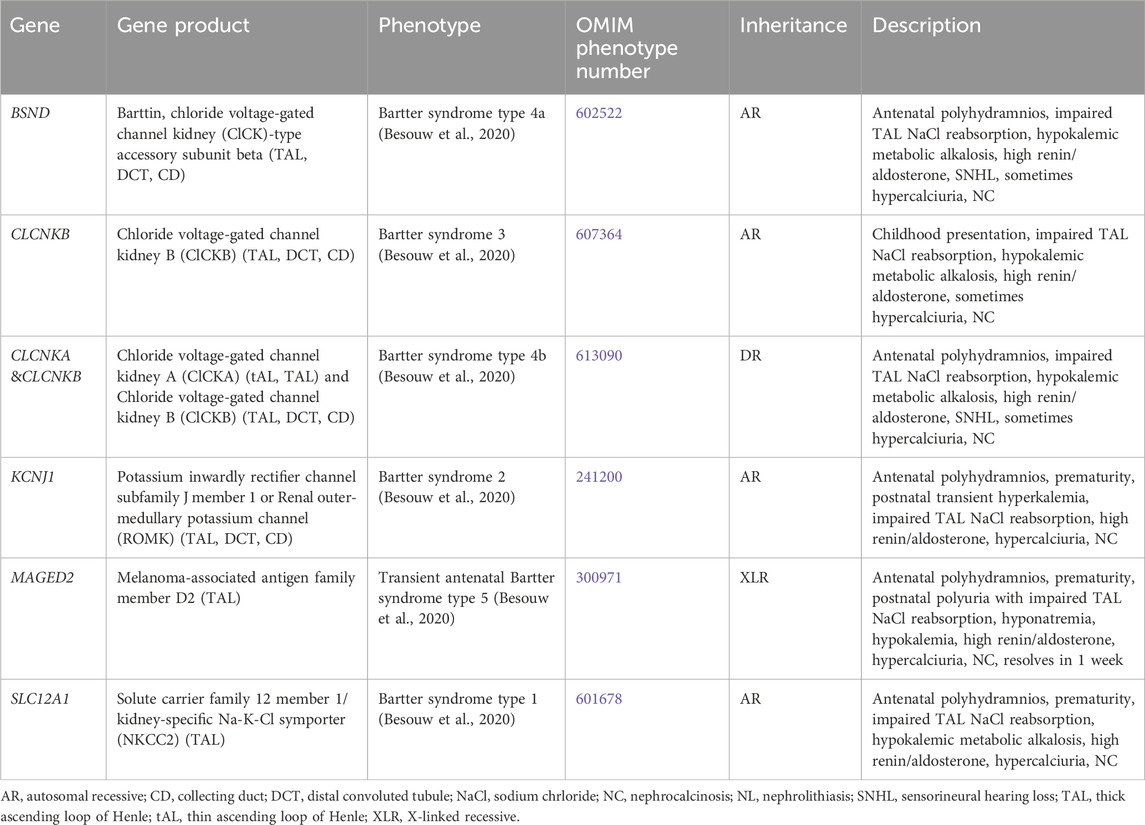
Table 7. Genetic causes of Bartter syndrome with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
Antenatal in presentation is present for Bartter syndrome type 1 (SLC12A1 gene, encodes for kidney-specific Na-K-Cl symporter [NKCC2], AR inheritance, OMIM phenotype number 601678) (Figure 1), Bartter syndrome type 2 (KCNJ1 gene, encodes for renal out-medullar potassium channel [ROMK], AR inheritance, OMIM phenotype number 241200) (Figure 1), Bartter syndrome type 4a (BSND gene, encodes for Barttin, AR inheritance, OMIM phenotype number 602522) (Figure 1), Bartter syndrome type 4b (CLCKA and CLCKB genes, encode for chloride voltage-gated channel kidney A [ClCKA] and chloride voltage-gated channel kidney B [ClCKB], digenic recessive [DR] inheritance, OMIM phenotype number 613090) (Figure 1), and Bartter syndrome type 5 (MAGED2 gene, XLR inheritance, OMIM phenotype number 300971) (Besouw et al., 2020). However, Bartter syndrome type 5 is transient and resolves after 1 week (Besouw et al., 2020). The only Bartter syndrome that presents in childhood is Bartter syndrome type 3 (CLCKB gene, encode for ClCKB, AR inheritance, OMIM phenotype number 607364) (Figure 1) (Besouw et al., 2020). Mainstays of treatment are hydration, sodium and potassium repletion, and nonsteroidal anti-inflammatory drugs (NSAIDs) that help to minimize urinary losses of electrolytes and water (Besouw et al., 2020).
3.1.8 Hyperaldosteronism and pseudohyperaldosteronismGenetic causes of hyperaldosteronism and pseudohyperaldosteronism with hypercalciuria and NL and/or NC are shown in Supplementary Table S3. Studies have shown that individuals with hyperaldosteronism have a higher incidence of NL compared to the general population (Chang et al., 2022). Hyperaldosteronism has also been associated with NC, thought to be related to factors including increased sodium excretion and resultant hypercalciuria, hyperphosphaturia, hypocitraturia, and hypokalemia-associated ammonia-medicated nephropathy (Mittal et al., 2015). Genetic hyperaldosteronism conditions associated with NL and/or NC are Familial hyperaldosteronism type I/Glucocorticoid-remediable aldosteronism (CYP11B1 gene, AD inheritance, OMIM phenotype number 103900), Familial hyperaldosteronism type II (CLCN2 gene, AD inheritance, OMIM phenotype number 605635), Familial hyperaldosteronism type III (KCNJ5 gene, AD inheritance, OMIM phenotype number 613677), and Familial hyperaldosteronism type IV (CACNA1H gene, AD inheritance, OMIM phenotype number 617027) (Mittal et al., 2015; Chang et al., 2022). Pseudohyperaldosteronism type IIB/Gordon syndrome (WNK4 gene, AD inheritance, OMIM phenotype number 614491) is associated with hypercalciuria and NL (Mabillard and Sayer, 2019).
3.1.9 Hyperparathyroidism and hypoparathyroidismGenetic causes of hyperparathyroidism and hypoparathyroidism with hypercalciuria and NL and/or NC are listed in Supplementary Table S4. Genetic causes of hyperparathyroidism with NL and/or NC are Familial primary hyperparathyroidism (CDC73 gene, AD inheritance, OMIM phenotype number 145000) and Hyperparathyroidism 4 (GCM2 gene, AD inheritance OMIM phenotype number 617343) (Lila et al., 2012; El Allali et al., 2021). Multiple endocrine neoplasia types I (131100), IIa (171400), and IV (610755) are also associated with hyperparathyroidism and NL and/or NC (Lila et al., 2012; El Allali et al., 2021). Hypoparathyroidism, sensorineural deafness, and renal dysplasia (GATA3 gene, AD inheritance OMIM phenotype number 146255) is associated with hypercalciuria and NC (Chenouard et al., 2013).
3.1.10 Other causes of hypercalciuriaSupplementary Table S5 lists other genetic causes of hypercalciuria and NL and/or NC. This includes Susceptibility to absorptive hypercalciuria/Familial idiopathic hypercalciuria (ADCY10 gene, AD inheritance, OMIM phenotype number 143870) (Reed et al., 2002), Hypophosphatasia (ALPL gene, AR and AD inheritance, OMIM phenotype numbers 241500, 241510, 146300) (Barvencik et al., 2011; Whyte et al., 2012; Rothenbuhler and Linglart, 2017), IMAGE syndrome (CDKN1C gene, AD inheritance, OMIM phenotype number 614732) (Balasubramanian et al., 2010), Beckwith-Wiedemann syndrome (CDKN1C, ICR1, KCNQ1OT1 genes, AD inheritance, OMIM phenotype number 130650) (Weksberg et al., 2010), Cystic fibrosis (CFTR gene, AR inheritance, OMIM phenotype number 219700) (Katz et al., 1988), Obstructive azoospermia with NL (CLDN2 gene, XLR inheritance, OMIM phenotype number 301060) (Askari et al., 2019), Amelogenesis imperfecta type IG/Enamel-renal syndrome (FAM20A gene, AR inheritance, OMIM phenotype number 204690) (Wang et al., 2013), Neurodevelopmental disorder with microcephaly, cataracts, and renal abnormalities (GEMIN4 gene, AR inheritance, OMIM phenotype number 617913) (Alazami et al., 2015), Somatic mosaic McCune-Albright syndrome (GNAS gene, mosaic inheritance, OMIM phenotype number 174800) (Kessel et al., 1992
留言 (0)