
記住我








 Figure
Figure Box 1
Box 1Pulmonary hypertension is a heterogeneous disease composed of a number of different disorders characterized by abnormally high pressures in the lung vasculature (Table 1).1 The causes of the abnormally high pressures vary, but the resulting symptomatology is largely the same.
TABLE 1. - Clinical classification of pulmonary hypertension types Reproduced with permission from Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913.1—PAH
1.1—Idiopathic PAH
1.2—Heritable PAH
1.3—Drug- and toxin-induced PAH
1.4—PAH associated with:
1.4.1—Connective tissue disease
1.4.2—HIV infection
1.4.3—Portal hypertension
1.4.4—Congenital heart disease
1.4.5—Schistosomiasis
1.5—PAH long-term responders to calcium-channel blockers
1.6—PAH with overt features of pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis
1.7—Persistent pulmonary hypertension of the newborn
2—Pulmonary hypertension caused by LHD
2.1—Pulmonary hypertension caused by heart failure with preserved ejection fraction
2.2—Pulmonary hypertension caused by heart failure with reduced ejection fraction
2.3—Valvular heart disease
2.4—Congenital or acquired cardiovascular conditions leading to postcapillary pulmonary hypertension
3—Pulmonary hypertension caused by CLD and/or hypoxia
3.1—Obstructive lung disease
3.2—Restrictive lung disease
3.3—Other lung diseases with mixed restrictive/obstructive patterns
3.4—Hypoxia without lung disease
3.5—Developmental lung disorders
4—Pulmonary hypertension caused by pulmonary artery obstructions
4.1—CTE-PH
4.2—Other pulmonary artery obstructions
5—Pulmonary hypertension with unclear and/or multifactorial mechanisms
5.1—Hematologic disorders
5.2—Systemic and metabolic disorders
5.3—Others
5.4—Complex congenital heart disease
Normal pulmonary arterial systolic pressure is between 15 and 30 mm Hg, and normal pulmonary arterial diastolic pressure ranges from 4 to 12 mm Hg; mean pulmonary arterial pressure (mPAP) values range from 9 to 18 mm Hg.2 The pulmonary vascular bed normally is low resistance, and can hold a patient's entire cardiac output at pressures 15% to 20% of the systemic circulation.2
Pulmonary hypertension is defined hemodynamically as an mPAP greater than 20 mm Hg.3 Abnormally high pressures in the pulmonary vasculature result from pulmonary vessel remodeling or increased downstream pressures.4 These physiologic changes lead to a decrease in the diffusing capacity of the lungs and, over time, the right ventricle enlarges to adapt to the increase in afterload.5 As a result, patients typically develop dyspnea, fatigue, exercise intolerance, and, in more advanced disease, symptoms of right-sided heart failure, systemic fluid overload, and eventually death.3
Most often, pulmonary hypertension is a complication of an underlying cardiovascular or pulmonary disease. Development is associated with worsening symptoms and reduced life expectancy independent of the underlying disease.6 From 2003 to 2020, about 126,526 people died in the United States from pulmonary hypertension; 66% were women and 34% men.7 Higher mortality also was observed in Black patients and those living in rural areas.7
The World Health Organization classifies pulmonary hypertension into five clinical groups: Group 1, pulmonary arterial hypertension (PAH); Group 2, pulmonary hypertension caused by left-sided heart disease (PH-LHD); Group 3, pulmonary hypertension caused by chronic lung disease (PH-CLD); Group 4, chronic thromboembolic pulmonary hypertension (CTE-PH); and Group 5, pulmonary hypertension with unclear and/or multifactorial mechanisms (Table 1).1 Grouping patients with similar pathologic findings, hemodynamic characteristics, and treatment lets clinicians communicate effectively about the different causes and standardize the diagnostic workup and treatment.1
 Box 2
Box 2Most forms of pulmonary hypertension are managed by treating the underlying disease. However, targeted therapeutic options are available for patients with PAH (Group 1). Surgical management can be an option for patients with PAH refractory to medical treatment and patients in Group 4 (CTE-PH).2 Without treatment, all forms of pulmonary hypertension cause progressive right heart failure, which often is fatal.
PATHOPHYSIOLOGY AND PREVALENCETo diagnose and treat pulmonary hypertension appropriately, clinicians must understand the distinctive features of the various clinical classifications in each of the five groups.
Group 1. PAH is characterized by precapillary pulmonary hypertension with resting mPAP greater than 20 mm Hg, pulmonary arterial wedge pressure (PAWP) of 15 mm Hg or less, and pulmonary vascular resistance (PVR) greater than 2 Wood units.1 Environmental and genetic insults are thought to initiate the disease.8 This process leads to increased PVR.9Although PAH is relatively rare, with a prevalence of 5 to 25 cases per 1 million population and an incidence of 2 to 5 cases per 1 million population per year, it has significantly higher morbidity and mortality than other forms of pulmonary hypertension.10,11 PAH most commonly is idiopathic (50% to 60% of cases).3 Historically, it was more common in young to middle-aged women without coexisting medical conditions, but now is more frequently diagnosed in patients ages 65 years and older with cardiovascular comorbidities and is diagnosed at about the same rates in men and women.3,12 Other causes include inherited genetic mutations, connective tissue disease, and drugs or toxins (Table 1).1 Clinicians can distinguish PAH from other classifications based on hemodynamic information revealed on echocardiogram.2
Group 2. PH-LHD is a form of postcapillary pulmonary hypertension defined by mPAP greater than 20 mm Hg at rest and PAWP greater than 15 mm Hg on right heart catheterization, and is the most common type of pulmonary hypertension.6 The two hemodynamic phenotypes are isolated postcapillary pulmonary hypertension and combined pre- and postcapillary pulmonary hypertension.6 Between 65% and 80% of cases of pulmonary hypertension are caused by LHD.13 Progressive increase in left atrial pressure occurs as a result of heart failure with reduced or preserved ejection fraction or valvular heart disease.14 Group 3. PH-CLD is a form of precapillary pulmonary hypertension with mPAP greater than 20 mm Hg at rest, PAWP of 15 mm Hg or less, and PVR of 3 Wood units or greater on right heart catheterization.6 Causes include chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD), and other conditions that cause chronic hypoxia, such as obesity hypoventilation syndrome or overlap syndrome (a combination of obstructive sleep apnea and COPD).6 Hypoxia induces vasoconstriction in the lungs, redirecting blood flow to areas of higher oxygenation for more effective gas exchange. Chronic hypoxia causes widespread vasoconstriction of the lung vasculature, which over time leads to vascular remodeling; this in turn causes increased PVR.15The prevalence of pulmonary hypertension in patients with COPD varies depending on the patient's stage of COPD, the kind of diagnostic tool used, and the definition used for pulmonary hypertension.15 The key point for clinicians is not to underestimate the likelihood that most patients with COPD will develop pulmonary hypertension.15 Between 8% and 15% of patients with idiopathic pulmonary fibrosis (IPF) have pulmonary hypertension at initial diagnosis; this figure rises to 30% to 50% of patients with advanced IPF and more than 60% of those with end-stage IPF.15
Group 4. CTE-PH is a form of precapillary pulmonary hypertension with mPAP greater than 20 mm Hg at rest, PAWP of 15 mm Hg or less, and PVR of 3 Wood units or greater on right heart catheterization.6 Subgroups are based on the underlying cause. This rare form of pulmonary hypertension is caused by increased clotting and persistent thrombi following pulmonary embolisms after at least 3 months of therapeutic anticoagulation.3 The clots obstruct the pulmonary arteries, causing increased pressure in the lungs and diminished blood flow, leading to hypoxia and pulmonary vascular remodeling.16 The estimated incidence of CTE-PH is 0.9 patients per million and the estimated prevalence is 14.5 to 144 patients per million adults.11 Group 5. Pulmonary hypertension associated with one of several complex disorders and multiple etiologic factors may be secondary to increased pre- or postcapillary pulmonary hypertension or direct effects on pulmonary vasculature.6 Precapillary hypertension is characterized by mPAP greater than 20, PAWP of 15 or less, and PVR greater than 2 Wood units; postcapillary hypertension is characterized by mPAP greater than 20, PAWP greater than 15, and PVR less than 2 Wood units.6 Causes include hematologic disorders, metabolic disorders, chronic renal failure, and complex congenital heart disease.4 The incidence and prevalence of pulmonary hypertension in most of these disorders is unknown.3 CLINICAL MANIFESTATIONSProgressive dyspnea is the most common symptom of pulmonary hypertension, occurring as an initial symptom in more than half of patients and eventually present in about 85%.2 A high index of suspicion is needed to diagnose pulmonary hypertension; thus, clinicians should consider it in patients with exertional dyspnea. Studies have estimated that 21% of patients experienced a 2-year delay in receiving a diagnosis of pulmonary hypertension after symptoms began.4 Other symptoms include fatigue, chest pain, presyncope or syncope, lower extremity edema, and palpitations.2
As the disease progresses, symptoms progress to those of right-sided heart failure, including weight gain from fluid retention, right upper quadrant abdominal pain from hepatic congestion, and other signs of systemic venous congestion.
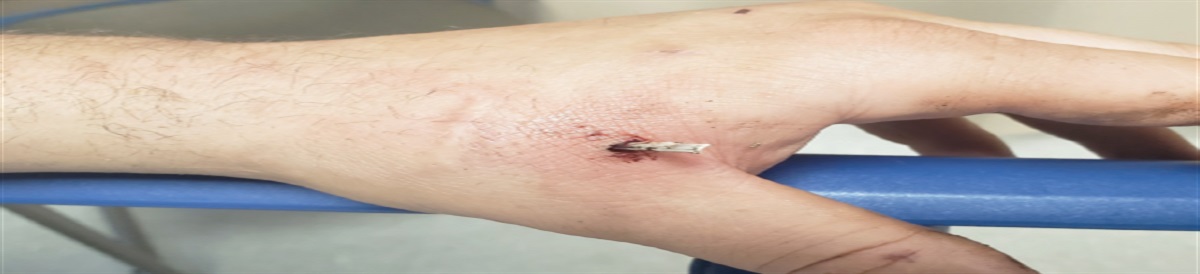
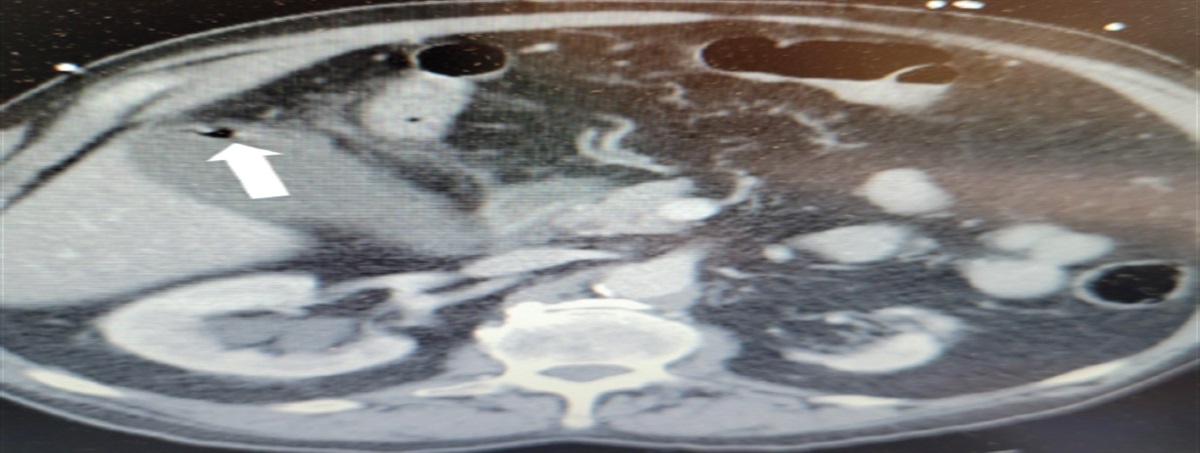
Poch and Mandel note that physical examination findings may be unremarkable in patients in the early stages of pulmonary hypertension, but as the disease progresses, findings suggestive of pulmonary hypertension include right-sided heart failure and systemic fluid overload.2 Signs may include jugular venous distension, hepatojugular reflux, hepatomegaly, ascites, peripheral edema, left parasternal heave, and S3 gallop. Less common symptoms include exertional chest pain, wheezing, cough, atelectasis, and Ortner syndrome (Figure 1).17
 FIGURE 1.:
FIGURE 1.: Ortner syndrome, hoarseness of the voice from compression of the left laryngeal nerve by an enlarged pulmonary arteryReproduced with permission from Kheok SW, Salkade PR, Bangaragiri A, et al. Cardiovascular hoarseness (Ortner's syndrome): a pictorial review. Curr Probl Diagn Radiol. 2021;50(5):749-754.
The combination of peripheral edema, right ventricular heave, and elevated jugular venous pressure highly suggests the presence of severely elevated mPAP and requires urgent diagnostic evaluation.18 Physical examination findings such as crackles, wheezing, heart murmurs, peripheral cyanosis, and other signs of left heart failure are more suggestive of PH-LHD or PH-CLD.2 When evaluating a patient with similar signs and symptoms, such as patients with left-sided heart failure, valvular heart disease, or obstructive or restrictive lung diseases, clinicians should keep pulmonary hypertension on the differential.
DIAGNOSISThe wide range of underlying disorders and conditions that can lead to pulmonary hypertension can complicate its diagnosis. In patients presenting with symptoms and physical examination findings consistent with pulmonary hypertension, common tests include ECG, chest radiograph, and pulmonary function tests (PFTs). Transthoracic echocardiography also can be useful to estimate the probability of pulmonary hypertension.19 The role of diagnostic testing is not only to confirm the presence of pulmonary hypertension, but to help identify the underlying cause and classify it. By classifying patients with PAH, CTE-PH, or other forms of severe pulmonary hypertension, clinicians can appropriately refer them to pulmonary hypertension centers for specialized care.20
In 2022, the European Society of Cardiology (ESC)/the European Respiratory Society (ERS) issued guidelines for the diagnosis and treatment of pulmonary hypertension.3 If a clinician has a patient with unexplained dyspnea or is suspected of having pulmonary hypertension, the clinician can refer to a diagnostic algorithm to approach the clinical workup (Figure 2).3 The algorithm has three steps:
SuspicionPatients are likely to present to their primary care providers (PCPs) with nonspecific cardiovascular or pulmonary symptoms. Perform a comprehensive workup including past medical and family history, physical examination, and bloodwork to narrow the differential diagnosis and point the clinician toward a cardiovascular or pulmonary cause.
 FIGURE 2.:
FIGURE 2.: Diagnostic algorithm of patients with unexplained exertional dyspnea and/or suspected pulmonary hypertension.Reproduced with permission from Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-3731.
DetectionNoninvasive cardiac and/or lung testing should be performed next based on the clinician's suspicion of the underlying cause of the symptoms. These tests may include an ECG, chest radiograph, PFTs, arterial blood gas (ABG) analysis, and echocardiogram. Of these tests, the echocardiogram is the most helpful, because it assesses right and left heart morphology and the level of probability of pulmonary hypertension, irrespective of the cause. This information also helps guide subsequent testing for the classification of pulmonary hypertension and determines whether invasive right heart catheterization is warranted.3
Guidelines recommend using the peak tricuspid regurgitation velocity measured by echocardiography as the key variable for assessing the probability of pulmonary hypertension.3 However, the presence or absence of pulmonary hypertension cannot be reliably determined by tricuspid regurgitation velocity alone. Peak tricuspid regurgitation velocity is used to separate patients into risk groups: low (2.9 m/s or less), intermediate (greater than 2.9 m/s to 3.4 m/s), and high (greater than 3.4 m/s).21 Additional echocardiography findings are used to further evaluate a patient's probability of having pulmonary hypertension. Other findings suggestive of pulmonary hypertension include an elevated pulmonary artery systolic pressure, right or left ventricular enlargement, flattening of the interventricular septum, pulmonic insufficiency, midsystolic notching, an enlarged pulmonary artery diameter, and an enlarged inferior vena cava with decreased inspiratory collapse.16
ConfirmationWhen echocardiography is highly suggestive of pulmonary hypertension, further diagnostic testing is warranted. Clinicians should focus on ruling out preexisting cardiovascular or pulmonary disease because these are the most common causes of pulmonary hypertension.6 If the patient has clear echocardiographic findings of LHD (for example, aortic valve disease, mitral valve disease, or heart failure with reduced or preserved ejection fraction in conjunction with evidence of pulmonary hypertension), further testing usually is not required to confirm the diagnosis.22 If uncertainty remains, consider right and/or left heart catheterization.14
Patients without evidence of LHD on echocardiography should undergo additional testing to identify noncardiac causes of pulmonary hypertension.3,13 The most common noncardiac causes of pulmonary hypertension are CLD and CTE-PH. Based on the patient's clinical presentation and reported history, clinicians should order additional testing. In patients with evidence of CLD, echocardiographic findings should be interpreted in conjunction with ABG analysis, PFTs, diffusing capacity of lungs for carbon monoxide, high-resolution chest CT, and a 6-minute walk test.3 Patients with evidence or history of thromboembolic disease should undergo ventilation-perfusion scanning to evaluate for CTE-PH.16
After the noninvasive workup has concluded, if the evidence suggests that the patient may indeed have pulmonary hypertension, the gold standard for diagnosis is right heart catheterization.3 According to Poch and Mandel, right heart catheterization is mandatory to establish a diagnosis of PAH and must be performed before advanced therapies are directed to the pulmonary vasculature.2
Right heart catheterization provides information on cardiopulmonary hemodynamics, including estimates of pulmonary arterial pressure, right ventricular pressure, and right atrial pressures. A pulmonary capillary wedge pressure (PCWP) of 15 mm Hg or less signifies the absence of pulmonary venous hypertension, which differentiates PAH from other causes of pulmonary hypertension.3 In addition, a PCWP of 15 mm Hg or greater signifies elevated left atrial pressure from LHD, which is only seen in patients with Group 2 pulmonary hypertension.
Treatment for pulmonary hypertension is based on the underlying pathology, with the 2022 ESC/ERS guidelines as a framework for recommended therapies.3
TREATING GROUP 1PAH-specific drug therapies should be prescribed by PAH specialists. PAH treatment involves FDA-approved medications in three major categories: endothelin receptor antagonists, phosphodiesterase (PDE) inhibitors and soluble guanylate cyclase (sGC) stimulators, and prostacyclins.23,24 These therapies enhance exercise capacity, improve quality of life, and slow disease progression. Each of the therapies, with a goal of lowering pulmonary vascular pressure, carries the risk of common adverse reactions such as systemic arterial hypotension, gastrointestinal (GI) intolerance such as nausea and vomiting related to dilation of the GI vascular bed, headache, and flushing.
What makes these therapies different is that they target unhealthy pulmonary vasculature; traditional vasodilators also affect healthy pulmonary vasculature.25 Before the development of targeted therapies, patients with PAH had a 1-year survival rate of 69% and a 5-year survival rate of 38%.26 Since development of targeted therapies over the past 2 decades, the rate for 1-year survival has increased to 85% and for 5-year survival to 57%.27 Research has borne out that patients who receive treatment within 6 months of diagnosis have a more pronounced effect and better prognosis.28
However, these therapies are costly and may only be available at certain medical centers. A review by Kaiser Permanente Colorado found that for a patient with PAH, the median total expenditure per day was $56, and 3-year total expenditures were $50,599.23 Data from the Pulmonary Hypertension Association Registry from 2015 to 2020 found that Black race, public insurance, lower level of education, low household income, higher number of people in household, high body mass index, previous drug use, older age, and male sex are associated with an increased risk of death, hospitalization, and clinical decline.29 Although targeted therapies offer promise, room for improvement remains in terms of treatment costs and social determinants of health.
Outside of the aforementioned targeted therapies, a subset of Group 1 patients may demonstrate vasoreactivity to oral calcium channel antagonist therapy. The target dosages for patients with PAH are two to three times the dosages typically used for patients with systemic arterial hypertension, which may concern PCPs unfamiliar with this higher dosing.3
Endothelin receptor antagonistsBosentan, ambrisentan, and macitentan block the action of endothelin, a peptide that constricts blood vessels. Adverse reactions include peripheral edema and hepatotoxicity. These drugs are teratogenic and should not be used in pregnant patients.3
PDE inhibitors and sGC stimulatorsThese therapies are linked to the nitric oxide pathway. Nitric oxide is a signaling molecule that acts as a vasodilator by binding to sGC, which catalyzes the conversion of guanosine triphosphate to cGMP, an intracellular messenger that relaxes smooth muscle. PDE is an enzyme that breaks down cGMP. Sildenafil and tadalafil inhibit the action of PDE-5, promoting vasodilation. Adverse reactions include visual or auditory disturbances.30 Riociguat stimulates sGC, leading to increased production of cGMP and vasodilation even in the absence of nitric oxide.30
Riociguat and all of the endothelin receptor antagonists are teratogenic and highly regulated by the FDA.31 The Risk Evaluation and Mitigation Strategy (REMS) is a required risk management plan determined by the FDA that includes special requirements or certifications for prescribers, dispensing pharmacists, documentation of patient safety monitoring such as laboratory tests, and patient enrollment in a registry.32 Riociguat, for example, is available to female patients only through a REMS program.
Prostacyclin analogs and receptor agonistsEpoprostenol, treprostinil, and iloprost are prostacyclin analogs and mimic the effects of prostacyclin, a naturally occurring substance that dilates blood vessels and inhibits platelet aggregation.30 Selexipag, a prostacyclin receptor agonist, selectively activates prostacyclin receptors.30 Common adverse reactions include headache, jaw pain (especially with treprostinil), flushing, and diarrhea.
Combination therapiesSome patients may receive a combination of the above medications for a more comprehensive approach to managing pulmonary hypertension. However, avoid any combination of PDE inhibitors, sGC stimulators, and nitrates because of the risk for severe hypotension.3 Patients with PAH also should be immunized against COVID-19, influenza, and Streptococcus pneumoniae.3
TREATING GROUP 2Pulmonary hypertension secondary to heart failure should be managed with diuretics.3 Use of medications approved for PAH in patients from Group 2 may have detrimental effects on gas exchange or hemodynamics and is not recommended.3 Patients should be immunized against COVID-19, influenza, and S. pneumoniae.33
TREATING GROUP 3Patients with pulmonary hypertension caused by lung pathologies and/or hypoxia may benefit from supplemental oxygen, noninvasive ventilation, and pulmonary rehabilitation programs.3 Use of medications approved for PAH in patients with Group 3 pulmonary hypertension may have detrimental effects on gas exchange or hemodynamics. Inhaled treprostinil has FDA approval for treatment of ILD, but the ESC/ERS guidelines suggest only that its use may be considered without providing a strong recommendation.3,34 Patients with COPD should be immunized against COVID-19, influenza, and S. pneumoniae.35
TREATING GROUP 4Treatment includes a combination of surgical pulmonary endarterectomy, balloon pulmonary angioplasty, and medical therapies. Lifelong therapeutic anticoagulation is recommended. Riociguat is recommended for symptomatic patients with CTE-PH or those with persistent pulmonary hypertension after pulmonary endarterectomy.3
TREATING GROUP 5No PAH therapies have demonstrated benefit or are recommended for patients in this group. Therapies are confined to treating the underlying disorder.3
CONCLUSIONDespite advances in understanding and treating pulmonary hypertension, the disease continues to be associated with high morbidity and mortality. The prevalence of pulmonary hypertension should make clinicians concerned and interested in identifying these patients. Awareness of the condition by PCPs is important because they may be the first to recognize signs and symptoms, leading to diagnosis and treatment.
REFERENCES 1. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. 2. Poch D, Mandel J. Pulmonary hypertension. Ann Intern Med. 2021;174(4):ITC49–ITC64. 3. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731. 4. Sarah B, Ashrith G, Sandeep S. Evaluation, diagnosis, and classification of pulmonary hypertension. Methodist DeBakey Cardiovasc J. 2021;17(2):86–91. 5. Fayyaz AU, Edwards WD, Maleszewski JJ, et al. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. 2018;137(17):1796–1810. 6. Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4(4):306–322. 7. Singh H, Agarwal L, Jani C, et al. Pulmonary hypertension associated mortality in the United States from 2003 to 2020: an observational analysis of time trends and disparities. J Thorac Dis. 2023;15(6):3256–3272. 8. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122(12):4306–4313. 9. Nie L, Li J, Zhang S, et al. Correlation between right ventricular–pulmonary artery coupling and the prognosis of patients with pulmonary arterial hypertension. Medicine (Baltimore). 2019;98(40):e17369. 10. Maron BA, Galiè N. Diagnosis, treatment, and clinical management of pulmonary arterial hypertension in the contemporary era: a review. JAMA Cardiol. 2016;1(9):1056–1065. 11. Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ. 2021;11(1):2045894020977300. 12. Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14(10):603–614. 13. Rosenkranz S, Gibbs JSR, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016;37(12):942–954. 14. Vachiéry J-L, Tedford RJ, Rosenkranz S, et al. Pulmonary hypertension due to left heart disease. Eur Respir J. 2019;53(1):1801897. 15. Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53(1):1801914. 16. Remy-Jardin M, Ryerson CJ, Schiebler ML, et al. Imaging of pulmonary hypertension in adults: a position paper from the Fleischner Society. Radiology. 2021;298(3):531–549. 17. Kheok SW, Salkade PR, Bangaragiri A, et al. Cardiovascular hoarseness (Ortner's syndrome): a pictorial review. Curr Probl Diagn Radiol. 2021;50(5):749–754. 18. Braganza M, Shaw J, Solverson K, et al. A prospective evaluation of the diagnostic accuracy of the physical examination for pulmonary hypertension. Chest. 2019;155(5):982–990. 19. Mandras SA, Mehta HS, Vaidya A. Pulmonary hypertension: a brief guide for clinicians. Mayo Clin Proc. 2020;95(9):1978–1988. 20. Sirajuddin A, Mirmomen SM, Henry TS, et al. ACR appropriateness criteria suspected pulmonary hypertension: 2022 update. J Am Coll Radiol. 2022;19(11):S502–S512. 21. Gall H, Yogeswaran A, Fuge J, et al. Validity of echocardiographic tricuspid regurgitation gradient to screen for new definition of pulmonary hypertension. EClinicalMedicine. 2021;34:100822. 22. Kanwar MK, Tedford RJ, Thenappan T, et al. Elevated pulmonary pressure noted on echocardiogram: a simplified approach to next steps. J Am Heart Assoc. 2021;10(7):e017684. 23. Burger CD, Ghandour M, Padmanabhan Menon D, et al. Early intervention in the management of pulmonary arterial hypertension: clinical and economic outcomes. Clinicoecon Outcomes Res. 2017;9:731–739. 24. Galiè N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. 25. Enderby CY, Burger C. Medical treatment update on pulmonary arterial hypertension. Ther Adv Chronic Dis. 2015;6(5):264–272. 26. D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. 27. Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. 28. Gaine S, Sitbon O, Channick RN, et al. Relationship between time from diagnosis and morbidity/mortality in pulmonary arterial hypertension: results from the phase III GRIPHON study. Chest. 2021;160(1):277–286. 29. Grinnan D, Kang L, DeWilde C, et al. Prediction of patient outcomes through social determinants of health: the Pulmonary Hypertension Association Registry (PHAR) evaluation. Pulm Circ. 2022;12(3):e12120. 30. Zolty R. Pulmonary arterial hypertension specific therapy: the old and the new. Pharmacol Ther. 2020;214:107576. 31. US Food and Drug Administration. Risk evaluation and mitigation strategies (REMS). www.accessdata.fda.gov/scripts/cder/rems/index.cfm. Accessed January 23, 2024. 32. US Food and Drug Administration. FDA's application of statutory factors in determining when a REMS is necessary. www.fda.gov/regulatory-information/search-fda-guidance-documents/fdas-application-statutory-factors-determining-when-rems-necessary. Accessed January 23, 2024. 33. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;79(17):e263–e421. 34. Waxman A, Restrepo-Jaramillo R, Thenappan T, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–334. 35. Agustí A, Celli BR, Criner GJ, et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD executive summary. Am J Respir Crit Care Med. 2023;207(7):819–837.
留言 (0)