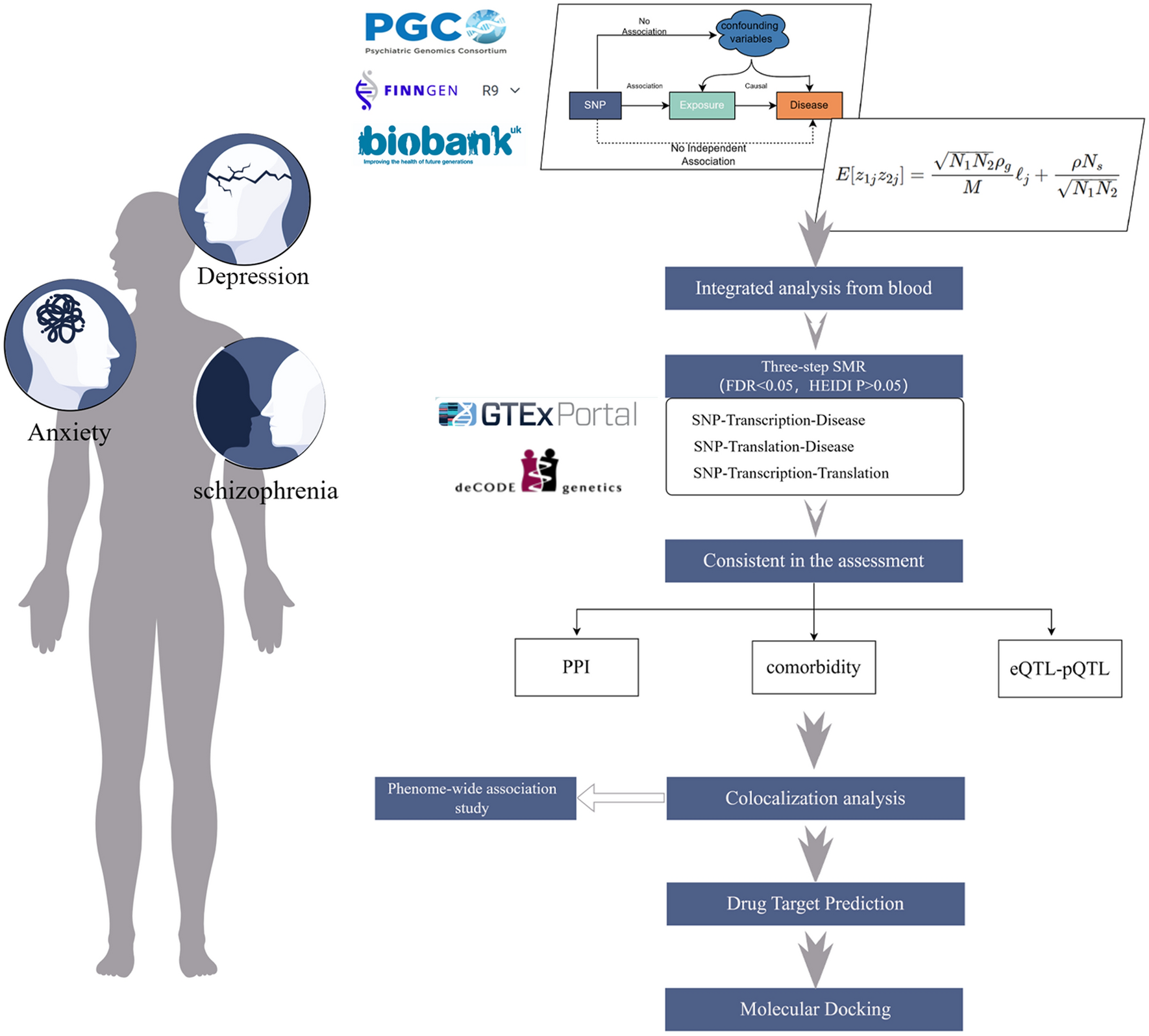
Gaining an in-depth understanding of the intricate relationship between genetic discoveries and pharmaceutical targets holds paramount importance in effectively translating GWAS findings into clinical applications [32]. Our study assumes pioneering significance as the first to center on actionable druggable genes, unlocking the potential of drug repurposing strategies within the domain of MG treatment. Notably, we identified three genes (CDC42BPB, CD226, and PRSS36) and three proteins (PRSS8, CTSH, and CPN2) as having significant MR results using cis-eQTL and cis-pQTL genetic instruments, which also exhibit compelling evidence of colocalization with MG. Additionally, through a meticulous exploration of immune cell-specific effects, this study may shed light on mechanistic insights underlying the loci associated with MG.
Our MR analysis establishes a compelling link between genetic variants associated with increased CDC42BPB gene expression and an elevated MG susceptibility. This finding underscores the potential of CDC42BPB inhibitors as a promising avenue for therapeutic intervention. CDC42BPB is a serine/threonine protein kinase intricately involved in regulating actin cytoskeleton dynamics and cell contraction. Several small molecules or biological inhibitors of CDC42BPB, such as SR-7826, BDP8900 and BDP9066 [33], have exhibited antitumor activity. However, a comprehensive understanding of the mechanistic underpinnings through which elevated CDC42BPB contributes to the pathophysiology of MG necessitates further investigation. Conversely, we observed a protective effect associated with increased CD226 gene expression against MG. CD226, also known as DNAX accessory molecule-1 (DNAM-1), is a member of the immunoglobulin superfamily and is expressed on various immune cells, including natural killer (NK) and T cells [34]. CD226, along with the inhibitory receptors TIGIT and CD96, constitutes cell-surface receptor family 3, binding to nectin and nectin-like proteins [35]. Prior research emphasizes the potent roles of this receptor family in regulating tumor immunity [36]. These findings underscore the importance of maintaining the expression of the activating receptor CD226, in orchestrating effective immune responses. Genetic polymorphisms within the CD226 gene have been associated with serval autoimmune diseases, including multiple sclerosis [37]. Building on these insights, we hypothesize that targeted manipulation of CDC42BPB and CD226 expression could potentially serve as a potent therapeutic strategy for managing MG. Nonetheless, a comprehensive validation of these proposed therapeutic targets necessitates further rigorous investigation.
Our study emphasizes a consistent and robust association between genetically predicted circulating Cathepsin H abundance and an elevated risk of MG across diverse datasets, including the UK Biobank and FinnGen Biobank. The subgroup analyses further revealed nuanced insights, particularly the association between genetically predicted circulating CTSH abundance and an increased risk of late-onset MG. Additionally, the posterior probability that CTSH expression levels in TH2 cells and MG outcomes shared a single causal signal in the 1-Mb locus around the cis-pQTL, rs12148472, was 0.8854 for MG susceptibility (Additional file 1: Figure S5). This observation implies that CTSH abundance might serve as a biomarker or contribute to the biological mechanisms underlying MG. CTSH encodes cathepsin H, a member of the papain-like cysteine proteases that are involved in major histocompatibility complex class II antigen presentation. Notably, CTSH has garnered attention for its involvement in type 1 diabetes [38] and narcolepsy risk [39], both of which prominently feature autoimmune components. Worth noting is the fact that other members of the cathepsin family, such as cathepsin S and cathepsin K, have received more attention as drug targets for autoimmune diseases. For example, cathepsin S inhibitors have been investigated for their potential in treating diseases like and multiple sclerosis [40].
Furthermore, our study provided robust genetic evidence supporting a potential causal role of increased CPN2 protein levels in reducing the risk of MG. CPN2, also known as Carboxypeptidase N Subunit 2, forms a complex with enzymatically active small subunits (CPN1). CPN plays a pivotal role as a zinc metalloprotease responsible for cleaving and partially inactivating anaphylactic peptides, specifically complement component 3a (C3a) and C5a, within the classical and lectin pathways of complement activation [41]. Protein–protein interaction studies using the STRING database (https://string-db.org/) have shown interactions between CPN2 and C5 (Additional file 1: Figure S6). It is widely recognized that dysregulated complement activation is a primary pathogenic mechanism in MG. Notably, C5 inhibitors, such as Eculizumab, have proven effective in preventing complement-dependent membrane attacks at the neuromuscular junction, presenting a promising avenue for the treatment of MG [42]. Future research is warranted to investigate the potential therapeutic implications of CPN2 in MG.
A significant hurdle in our analytical pursuit lay in discerning whether the association with MG risk stemmed from PRSS36, PRSS8, or both entities, given the substantial correlation between the cis-eQTL instrument for PRSS36 (rs78924645) and cis-pQTL instrument (rs1060506) for PRSS8 (r2 = 0.93 in 1000 Genomes Project European ancestry participants). Considering that both PRSS36 and PRSS8 belong to the serine protease family, a group of proteolytic enzymes involved in diverse biological processes, we regard them as potential drug targets for MG. It is worth noting that PRSS8 inhibits TLR4-mediated inflammation in human and mouse models of inflammatory bowel disease [43], thereby implicating its plausible relevance to TLR4-mediated inflammation pathways in the context of MG. As such, the identification of PRSS36 and PRSS8 as putative targets holds promise; however, the trajectory towards their validation as credible therapeutic avenues warrants further inquiry, coupled with a comprehensive assessment of their viability for subsequent drug development endeavors.
Genetic variation controls transcriptional regulation in a cell type-specific manner to regulate immune pathways [44, 45]. The cell-type specificity of the effects elicited by common genetic variants on both gene and protein expression engenders a pronounced reliance on the precise cellular contexts. In navigating this intricacy, the relationships underpinning eQTL and pQTL manifest a profound dependence on the distinct cell types under scrutiny. In this study, we embarked on a comprehensive exploration, utilizing genetic colocalization analysis, to delineate the interplay between immune-cell-specific eQTLs and MG susceptibility, thereby unearthing the underlying molecular mechanisms. This approach yielded valuable insights into the genetic orchestration governing CTSH expression across an array of distinct cell types. Of note is the robust colocalization unveiled between CTSH expression within TH2 cells and the propensity for MG risk, signifying a putative involvement of CTSH expression within this cellular subtype in driving the genesis and advancement of MG. This revelatory aspect of our study underscores the imperative of acknowledging and integrating cell type heterogeneity within the realm of future research pursuits aimed at identifying potential therapeutic targets for MG. This newfound awareness stands poised to wield transformative influence in guiding the trajectory of future scientific explorations and therapeutic innovations targeting MG and other akin disorders.
The current study notably complements and extends previous efforts by employing key approaches to protect against potential biases, strengthen causal inference and enhance understanding of potential mechanisms. With our rigorous instrument selection process that used comprehensive datasets on gene expression and plasma protein levels, we have facilitated a thorough evaluation of actionable drug targets, notably including the previously unexplored CTSH. Our study, enriched with multi-omics data, has successfully overlooked targets that do not align with the potentially druggable gene targets prioritized in the Chia et al. study, as indicated by the Priority Index analysis [9, 46]. It is crucial to underscore that the peak cis-eQTL or cis-pQTL identified at each locus did not attain the genome-wide significance level threshold. Consequently, these loci were not deemed significantly associated with MG in Chia et al.'s study. This constitutes a noteworthy extension to previous study, emphasizing our pivotal role in contributing novel insights that surpass the confines of earlier investigations.
Several limitations need to be considered in our study. Firstly, only a small proportion of genes/proteins may be effectively instrumented by multiple SNPs, with the majority being instrumented by only two or one SNP. This limitation restricts our ability to conduct sensitivity analyses. Secondly, there is a potential concern related to the presence of epitope-binding artifacts caused by coding variants. These artifacts may introduce spurious signals, potentially leading to false-positive cis-pQTLs. Thirdly, our study is confined to AchR-positive MG cases, so caution is advised when extrapolating our results to patients with anti-MuSK and other autoantibodies. Additionally, although the GWAS summary data integrated into our study already constitute the most extensive MG GWAS dataset currently available, the limited number of cases in both the primary and replication datasets constrained our ability to replicate the MR estimates observed in the discovery cohort. Therefore, it is crucial to conduct further research with larger and more diverse populations, especially including non-European individuals and patients with anti-MuSK and other autoantibodies.





留言 (0)