
記住我




All experiments were performed according to Animal Use Guidelines and approved by the Ethics Committee of Ningxia Medical University (No.2022-206). Forty-eight male 6-week-old BALB/c mice (22±1g) were obtained from Beijing Huafukang Bio-Technology. Co., LTD (Beijing, China). Mice were fed in the Laboratory Animal Research Center of Ningxia Medical University (Yinchuan, China) under a 12 h light and dark cycle with free access to food and water. The mouse model of PNS was established by a single injection of doxorubicin hydrochloride (DOX; MCE, USA) 10mg/kg through the tail vein [20]. One week later, the mice were placed in a mice metabolic cage to collect 24 h urine and measure 24 h urine protein. After successful modeling, the subsequent experiments were performed.
Bacterial preparationC. butyricum was a vacuum freeze-dried strain provided by the China General Microbiological Culture Collection Center (CGMCC, strain number 1.5205). PYG MEDIUM (modified; Shandong, China) was used to resuscitate the bacteria. The bacteria were cultured in an anaerobic incubator (5% carbon dioxide) at 37 °C for about 24 h, and the fully grown bacteria were visible. The colonies were placed in 10% skim milk to make a freeze-dried powder and stored at -80 °C. During the intervention, PYG MEDIUM was used daily to resuscitate C. butyricum lyophilized powder in an anaerobic incubator (5% carbon dioxide) at 37 °C for 24 h, centrifuged at 3000×g for 5 min, and resuspended in sterile saline. The experimental final concentration was 1×108 CFU/mL.
Experimental designThe time diagram of the experimental design is shown in Fig. 1A. The mice were randomly divided into 4 groups (12 mice/group): CON, CON+C. butyricum, PNS, and PNS+C. butyricum. The CON+C. butyricum and PNS+C. butyricum groups were given 200μL C. butyricum by gavage once a day for 6 weeks. Meanwhile, mice in CON and PNS groups were treated with 200 μL normal saline at the same time. The 24 h urine was collected every 2 weeks using a mice metabolic cage, and the 24 h urine protein was measured by Bradford protein detection kit (Thermo Fisher, USA) to observe the changes of urine protein in mice. During the experiment, the body weights (BWs) of the mice were monitored weekly. After 6 weeks of gavage, fresh stool, and urine samples were collected and immediately frozen at -80 °C for subsequent analysis. At the termination of the experiment, mice were anesthetized with isoflurane inhalation (0.41 mL/min at 4 L/min fresh gas flow, application concentration 2%) was performed under the IACUC protocol and euthanized via transcardiac perfusion. The blood samples were taken from the orbit of mice and collected in tubes containing ethylenediaminetetraacetic acid (EDTA), and centrifuged at 4 °C (600×g for 10 min) to obtain plasma samples, which were stored at -80 °C for further study.
Fig. 1
The impacts of C. butyricum treatment on BWs, 24 h urinary protein and kidney function in DOX-induced PNS mice. Experimental design time diagram (A). BWs: Body weights (B). 24 h urine protein of mice in diverse groups (C). BUN: Blood urea nitrogen (D). SCr: Serum creatinine (E). UUN: Urine urea nitrogen (F). UCr: Urine creatinine (G). Data were expressed as mean±SD. *P < 0.05, **P < 0.01, *** P < 0.001, ****P < 0.0001. All experiments were performed in triplicate
Plasma and urine biochemistry testsBlood and urine samples were measured for the following biochemical properties: Blood urea nitrogen (BUN), serum creatinine (SCr), urine urea nitrogen (UUN), and urine creatinine (UCr). BUN and UUN were detected by urease method with a urea nitrogen test box; SCr and UCr were measured by creatinine assay kit using the sarcosine oxidase method (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Kidney tissue pathological section stainingAfter the sacrifice of the mice, the kidney tissues were immediately fixed in 4% paraformaldehyde, washed with tap water, and dehydrated with ethanol, paraffin-embedded sections (5μm) were placed on slides. Hematoxylin & eosin (HE) staining (Servicebio, NO.G1004, NO.G10001, Wuhan China), Masson staining (Solarbio, NO.G1340, Beijing, China), Periodic Acid Schiff (PAS) staining (Solarbio, NO.G1281, Beijing, China), and Periodic acid-silver methenamine (PASM) staining (Solarbio, NO.G1790, Beijing, China) were performed on the sections according to the standard protocols.
After completing the above staining steps, the sections were observed and photographed under a microscope (Aomori Olympus glass slide, Japan) at 40X. The kidney interstitial cell infiltration score was evaluated according to HE staining, semi-quantitative assessment of the degree of inflammatory cell infiltration by counting the percentage of inflammatory cell infiltration area and classified as follows: 0 (nil), 1 (<25%), 2 (25–50%), 3 (50–75%), and 4 (>75% of tubulointerstitial fields). Each score was determined by evaluating three sections per kidney and five non-overlapping visual fields per section [21]. Image J software (National Institutes of Health Bethesda, MD) was used to quantify the percentage of PAS-stained glycogen-covered area in the total image area and the percentage of kidney interstitial fibrosis area in the total image area. Observers were blinded to the experimental groups.
Determination of lipopolysaccharides (LPS) levels in kidney and gutThe Limulus amebocyte lysate kit (Xiamen Bioendo Technology Co., Ltd., Xiamen, China) was used to detect LPS levels in the intestine and kidneys of mice according to the manufacturer's instructions. Initially, the endotoxin standard solution was prepared according to the instructions. 100 μL of diluted intestine/kidney tissue homogenate supernatants (1:30/1:20 dilution with endotoxin-free water), endotoxin standard solution, and bacterial endotoxin test water were added into endotoxin-free test tube respectively. Bacterial endotoxin test water was used as a negative control, and endotoxin standard solution was used as a positive control. At the initial time point, 100μL of the Limulus amebocyte lysate agent was added to each tube and incubated at 37°C for 7 min. Then, 100 μL of chromogenic substrate was added to each tube and incubation at 37°C for 6 min. After the incubation, 500 μL of azo reagent 1, azo reagent 2, and azo reagent 3 were added in turn and allowed to stand for 5 min. The optical density was measured at 545 nm using a microplate reader (Thermo Scientific, Waltham, USA).
Enzyme-linked immunosorbent assay (ELISA)Kidney tissue homogenate supernatants were collected to determine the concentration of cytokines. Interleukin (IL)-6, IL-10, and IL-17A in the kidney were determined by using an ELISA kit (Proteintech, Wuhan, China) according to the manufacturer's instructions. The optical density at a wavelength of 450 nm was measured with an automatic microplate reader (Thermo Fisher Science Inc., USA).
Quantitative real-time PCRAccording to the manufacturer's protocol, total RNA was extracted from kidney tissue using the RNA Extraction kit (Omega, USA), and the UEIris RT mix with DNase (All-in-One) (UE, China) was used to synthesize cDNA. Then, RT-qPCR was performed using Universal SYBR Green qPCR Supermix (UE, China). The expression of the target gene was normalized by GAPDH. All experiments were carried out in three independent experiments. Primer sequences (Sangon Biotech, Shanghai, China) are shown in Table 1.
Table 1 List of primers used for qRT-PCRWestern blotThe kidney tissue protein was extracted using the whole protein extraction kit (Keygen, NO.KGP250, China) according to the manufacturer's protocol. Protein concentrations were detected with a BCA protein assay kit (Keygen, No.KGP902, China). 30 μg protein was added to each well of SDS-polyacrylamide gel (SDS-PAGE) for electrophoresis. The voltage of the concentrated gel was 90 V, and the voltage was changed to 120 V when the protein was transferred to the separation gel. After electrophoresis, the protein was transferred to the methanol-soaked polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, USA). Cell membranes were then blocked with 5% skim milk at room temperature for 2 h and incubated overnight at 4 ℃ with primary antibody including Heme oxygenase-1 (HO-1) rabbit mAb (1:5000 dilution, ABclonal, NO.A19062, China), signal transducer and activator of transcription 3 (STAT3) rabbit mAb (1:1000 dilution, ABclonal, NO.A19566, China), purified anti-retinoic acid-related orphan receptor gamma t (RORγt) Antibody (1:500, BioLegend, NO.603152, USA), Janus kinase 2 (JAK2) rabbit mAb (1:1000 dilution, CST, NO.3230S, USA), and mouse monoclonal GAPDH (1:5000 dilution, Proteintech, NO.60004-1-Ig, China). After washing with TBST buffer for 3 times, they were incubated with HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) (1:5000 dilution, Proteintech, NO.SA00001-2, China), HRP labeled Goat Anti-Mouse IgG (1:5000 dilution, Abbkine, NO.A21010, China), or HRP-conjugated Affinipure Goat Anti-Rat IgG (H+L) (1:5000 dilution, Proteintech, NO.SA00001-15, China) at room temperature for 1 h. After washing membranes with TBST buffer for 3 times, ECL chemiluminescence kit (Keygen NO.KGP1127, China) and SH-Compact 523 (Shenhua Technology Co., Ltd., China) were used for imaging and detection. Image J software (National Institutes of Health, Bethesda,USA) was used to analyze the gray value of the image.
Flow cytometry analysisThe cell suspension of the spleen was prepared. The spleen was ground first, centrifuged at 400×g, 4 ℃ for 5 min after passing through a 300-mesh filter membrane, and the supernatant was discarded. An appropriate amount of red blood cell lysate was added. After standing on ice for 5 min, the sample was centrifuged at 400×g for 5 min, the supernatant was discarded, and analyzed again according to the situation. Then RPMI 1640 medium was used to wash the cells. Finally, the cell concentration was adjusted to 1×106 cells/mL for subsequent detection.
Peripheral blood mononuclear cell (PBMC) suspension was prepared as described below. The peripheral blood of mice was collected in an anticoagulant tube containing EDTA, centrifuged at 600×g, 4 ℃ for 10 min and the plasma was frozen at -80 ℃. The remaining blood cell-containing liquid was transferred to the centrifuge tube and mixed with an appropriate amount of erythrocyte lysis solution. After standing on ice for 10 min, centrifuged at 4 ℃, 400×g for 5 min, discarded the supernatant, and lysed again according to the situation. The cells were washed with RPMI 1640 medium. Finally, the cell concentration was adjusted to 1×106 cells/mL for subsequent measurement.
The colon cell suspension was prepared. After the colon was removed, the colon was washed with normal saline, the adipose tissue and feces were removed, and the longitudinal dissection was performed. The intestinal segment was cut into about 1 cm, added with RPMI 1640 medium, and placed in a shaker at 37℃ for 30 min (shaken every 10 min for 10 s). After shaking for 15 s, the intestinal segment was allowed to sink for a moment to collect all the liquid, passing through a 200-mesh filter membrane (2 times) and collecting into a centrifuge tube. At the same time, a nylon wool column was prepared: glass wool was loosely filled in a 10 mL syringe and the column was firstly infiltrated with RPMI 1640 medium, the filtrate was continuously filtered 3 times. Then the collected intestinal cell fluid was filtered, and the column was washed with RPMI 1640 medium. All the liquid was collected and centrifuged for 500×g, 4 ℃ for 5 min, and the supernatant was discarded. The RPMI 1640 medium was added and centrifuged for 600×g, 4 ℃ for 20 min to collect the white membrane in the middle part. The RPMI 1640 medium was added and centrifuged for 400×g, 4 ℃ for 5 min. The supernatant was discarded and an appropriate amount of RPMI 1640 was added to resuspend the cells. The cell concentration was adjusted to 1×106 cells/mL for subsequent detection.
Flow cytometry was used to detect the percentages of Treg and Th17 cells in diverse tissues. For Treg cell staining, CD4-FITC (eBioscience, NO.2344796, USA) was used for surface labeling and eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher, USA) was elicited to fix and penetrate the cells. After that, the transcription factor forkhead box p3 (Foxp3)-PE (eBioscience, NO.4307350, USA) was added for intracellular labeling, and incubated at 4 °C for 30 min in the dark. For Th17 cell staining, cells were stimulated at 37 °C for 1 h using a 500× cell stimulation cocktail (Thermo Fisher, USA), followed by cell staining, including CD4-FITC (eBioscience, NO.2344796, USA) and IL-17A-PE (eBioscience, NO.4306419, USA), incubated at 4 °C in the dark for 30 min. Finally, prepared samples were measured and analyzed by Beckman Cyto FLEX flow cytometer (Beckman Bioscience, USA).
Gut and urethra microbiota analysisAfter 6 weeks of intervention with C. butyricum, 6 mice in each group were randomly selected to collect fresh feces in sterile cages and 3 mice in each group were randomly selected to collect fresh urine in sterile metabolic cages. The collected feces and urine were immediately stored at -80 ℃ until DNA was extracted.
The total DNA was extracted by Omega Mag-bind soil DNA kit (Omega M5636-02) and the DNA was quantified by Nanodrop, the quality of DNA extraction was detected by 1.2% agarose gel electrophoresis. The DNA in the sample was extracted using ultra-clean kits and reagents. UsingO5® High-Fidelity DNA Polymerase of TransGen Biotech (Beijing, China) with the primers 338F-5′ ACTCCTACGGGAGGCAGCAG 3′, 806R-5′ GGACTACHVGGGTWTCTAAT 3′ to amplified the hypervariable region V3-V4 of 16S rRNA sequence in bacterial DNA samples by PCR amplification instrument (ABI 2720, USA). A negative control during the PCR amplification of the target fragment. The negative control can detect microbial contamination such as environment and reagents, and any sample group with negative control amplification bands cannot be used for subsequent experiments. Quant-iT PicoGreen dsDNA Assay Kit fluorescence reagent and Microplate reader (BioTek, FLx800) quantitative instrument were used for fluorescence quantification of PCR amplification recovery products. The sequencing library was prepared using Illumina's TruSeq Nano DNA LT Library Prep Kit, and the library was subjected to final fragment selection and purification by 2% agarose gel electrophoresis. Finally, the sequencing was performed on a MiSeq sequencer by Suzhou Panomix Biomedical Technology Co., Ltd., China.
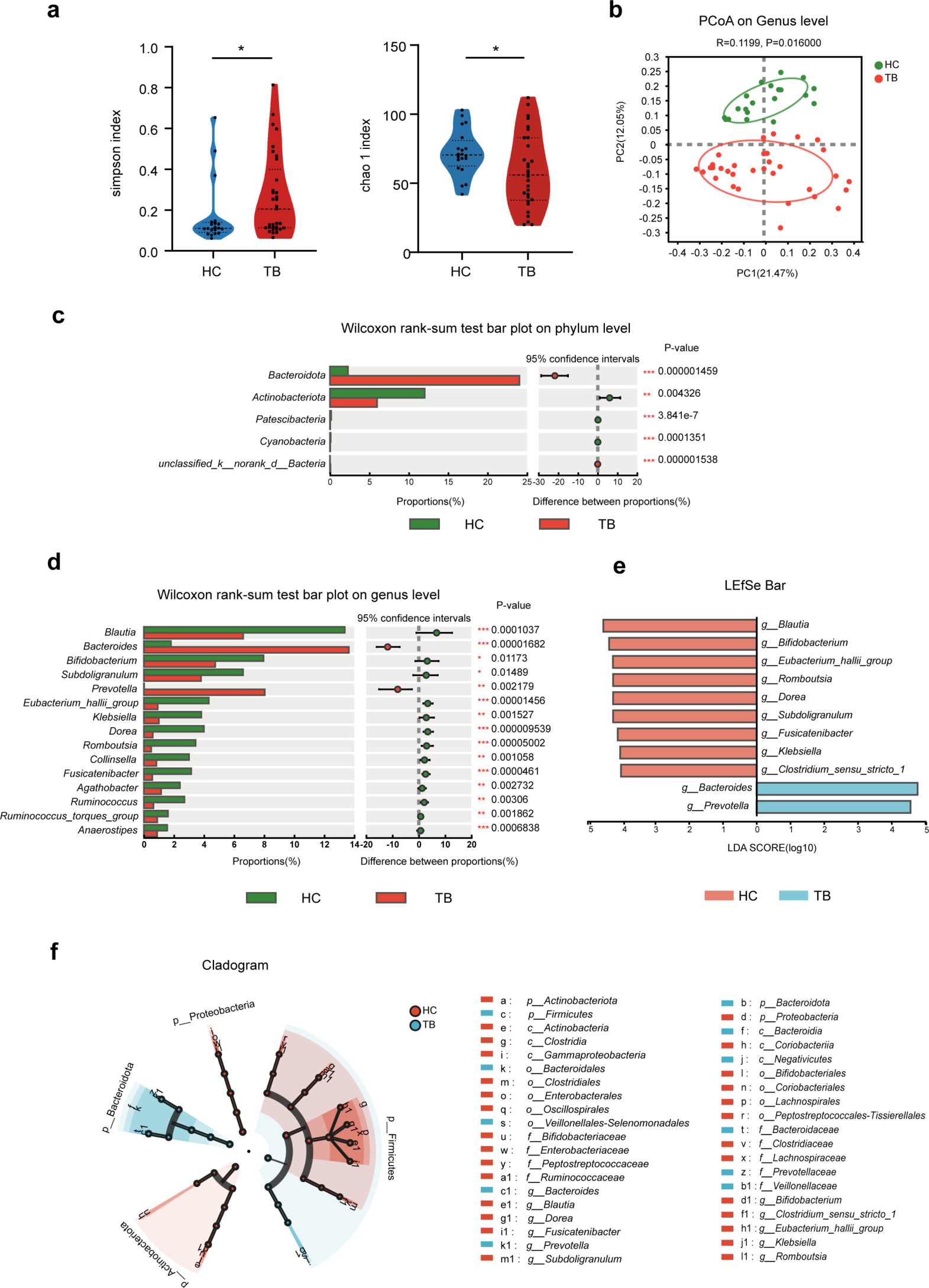
For the 16S rRNA gene of bacteria, Greengenes was selected as the reference database (Release 13.8). Using QIIME2 (2019.4) analysis software, the unmatched primer sequences were discarded first, and then the DADA2 plug-in was used for data processing such as quality filtering, denoising, splicing, and chimera removal. The obtained sequences were merged according to 100% sequence similarity to generate characteristic sequence ASVs and abundance data tables. Using the method of rarefaction, a certain number of sequences were randomly selected from each sample to reach a unified depth to predict the ASVs and their relative abundance that can be observed in each sample at the sequencing depth, which was used for subsequent species composition analysis, Alpha diversity analysis, Beta diversity analysis, and species difference analysis. Observed species, Chao1, and Shannon indices were used as measurements of alpha-diversity of the microbial communities. Jaccard-based principal coordinate analysis (PCoA) and nonmetric Multidimensional scaling (NMDS) analyses revealed beta diversity. For comparing the differences in species composition between samples, the Wayne diagram was used for community analysis, the heatmap was used for species composition analysis, and the PCA was used to analyze the differences in species composition between samples. LEfSe analysis was used to search for marker species between groups.
Measurement of the feces and urine SCFAs concentrationsEther was obtained from Titan (Shanghai, China). Phosphoric acid was obtained from Sinopharm (Shanghai, China). Acetic acid, propionic acid, isobutyric acid, butyric acid, isovaleric acid, valeric acid, 4-methylvaleric acid, and caproic acid were all obtained from Sigma-Aldrich (Shanghai, China). The 100 mg/mL stock solutions of 6 SCFAs (acetic acid, propionic acid, isobutyric acid, butyric acid, isovaleric acid, and valeric acid) and caproic acid were prepared by water and ether methods, respectively, and a series of working standard solutions were obtained by dilution.
Fecal standards were prepared, and metabolites were extracted. The internal standard (4-methylvaleric acid) was prepared with ether to 375 μg/mL, 200 μL series of working standard solutions of 6 acids, 100 μL 15 % phosphoric acid, 20 μL series of working standard solutions of hexanoic acid, 20 μL internal standard, and 260 μL either were mixed to prepare ten standard curve points, covering 0.02 to 500 μg/mL (0.02, 0.1, 0.5, 2, 10, 25, 50, 100, 250, 500 μg/mL). A total of 50 mg fecal samples were homogenized with 100 μL of 15% phosphoric acid, 20 μL of 375 μg/mL internal standard (4-methylpentanoic acid) solution, and 280 μL ether for 1 min, centrifuged at 4 °C 12000 rpm for 10 min. The supernatant was analyzed by gas chromatograph-mass spectrometric (GC-MS).
Urine standards were prepared, and metabolites were extracted. The internal standard (4-methylvaleric acid) was prepared with ether to 75 μg/mL. A series of working standard solutions of 200 μL six acids, 100 μL 15% phosphoric acid, 20 μL caproic acid, 20 μL internal standard, and 260 μL ether were mixed to prepare ten standard working solutions, covering from 0.02 to 100 μg/mL (0.02, 0.1, 0.5, 1, 2, 5, 10, 25, 50, 100 μg/mL). An appropriate amount of sample was taken in a 2 mL centrifuge tube, added with 50 μL 15% phosphoric acid, and then added with 10 μL of 75 μg/mL internal standard (isocaproic acid) solution and 140 μL of ether for homogenization for 1 min, centrifuged at 4 °C 12000 rpm for 10 min, and the supernatant was analyzed by GC-MS.
The GC analysis was performed on trace 1310 gas chromatograph. The chromatographic column was Agilent HP-INNOWAX capillary column (30 m×0.25 mm ID×0.25 μm) (Thermo Fisher Scientific, USA). MS detection of metabolites was performed on ISQ LT (Thermo Fisher Scientific, USA).
Statistical analysisGraphPad Prism version 9.0 (GraphPad Software Inc., La Jolla, CA, USA) and the statistical package for the social sciences (SPSS) 23.0 software (IBM Inc., Armonk, NY, USA) were used for statistical analyses. All experimental data were expressed as the mean±SD of at least three independent experiments. One-way analysis of variance was used to compare the mean values of variables among groups. After that, Tukey's post hoc test was used to determine the significance of pairwise comparison of mean values between groups. In addition, the expression correlation was analyzed by Spearman correlation coefficient assay. P < 0.05 was considered statistically significant.
留言 (0)