
記住我




Seventy-six participants were initially included in the microbiome metabolomics study, with 23 of them excluded from the microbiome analysis (12 sampling failures, 9 non-detects and 2 outliers) and 11 excluded from the metabolome analysis due to abnormal values. Paired microbiome and metabolomics samples from the remaining 42 participants (17 in the normal group and 25 in the HPV group) were subjected to microbiome and metabolome analyses. All HPV(+) subjects had infected one of 14 high-risk HPV genotypes: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66 and 68 (Supplementary Table 1). Seven of them had mixed infection of both low-risk and high-risk HPV (Supplementary Table 1). Analysis of clinical data from the 42 participants showed no statistical differences between the normal and HPV groups prior to collection (Supplementary Table 1), indicating that no knowable influencing factors affecting the analysis of the multi-omics study between the two groups were identified.
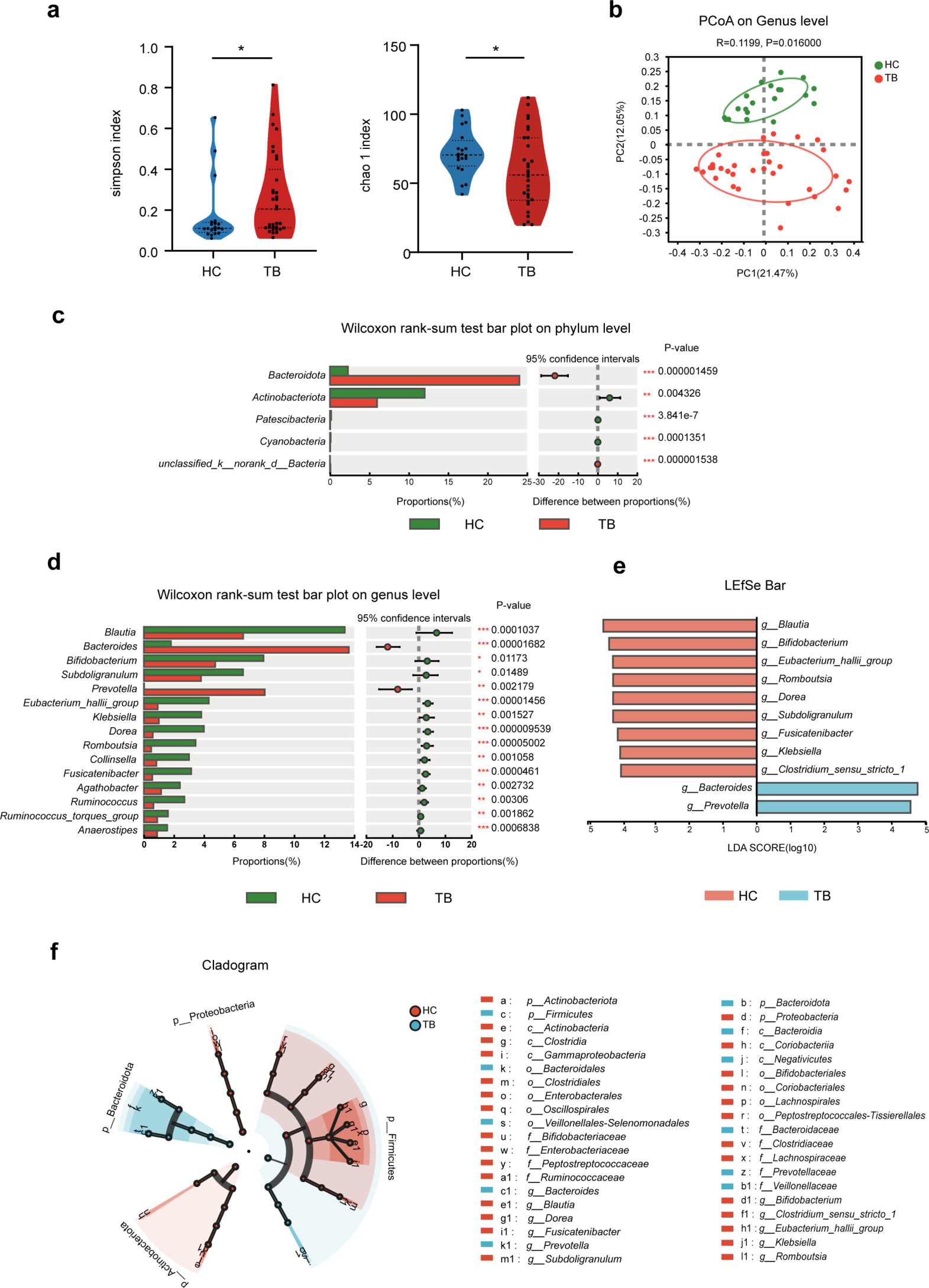
Cervicovaginal microbiome structure analysisAfter clustering the sequences into OTUs with 97% consistency by default, 625 OTUs were obtained (Supplementary Table 2). To assess the microbial structures, the alpha diversity (Observed species, Shannon, Simpson, Chao1 and Ace, Fig. 1A and E) and beta diversity (PCoA analysis; Fig. 1F and G) of the cervicovaginal microbiota was analyzed cross groups. Although we observed a slight increase of Shannon and Simpson indices in HPV group compared to the normal group, they together with other indices (Observed species, Chao1 and ACE) did not show statistical differences. Therefore, the alpha diversity was not statistically significant between the two groups. Based on the unweighted PCoA (Fig. 1F) and weighted PCoA (Fig. 1G) plots at OTU level, the bacterial structures in the two groups had some differences yet mostly were similar. The HPV group and normal group samples were not clearly separated (Fig. 1). These results suggest that HPV infection did not significantly alter the alpha diversity of cervicovaginal microbiota and may have an impact on specific microbial structures.
Fig. 1
Analysis of microbial diversity in cervix and vagina. A–E Species diversity difference between normal group and HPV group was estimated by Observed-species, Shannon, Simpson, Chao1 and Ace indices. HPV (n = 25), group with HPV infected patients; Normal (n = 17), subjects with normal cervical condition. F–G unweighted and weighted PCoA plot based on OTU levels showing bacterial structure clustering. HPV group (red), Normal group (blue)
Changes in the composition of the cervicovaginal microbiota associated with HPV infectionAnalysis of the top 10 species at the phylum level in the HPV and normal groups revealed significant differences in the cervicovaginal bacteria (Fig. 2). Firmicutes was the most dominant phylum, accounting for 36.07% of the HPV group and 28.03% of the normal group, respectively (Fig. 2A). The HPV group had higher levels of Actinobacteriota (18.17% vs. 2.84%), Fusobacteriota (5.70% vs. 0.26%), Cyanobacteria (0.21% vs. 0.26%) and Caldatribacteriota (0.038% vs. 0.004%) than the normal group. It is indicated that HPV remarkably impacts on cervicovaginal microbial abundance.
Fig. 2
A Relative abundance of the most abundant 10 phylum in the HPV group and Normal group. B–E statistically comparison of Firmicutes, Actinobacteriota, Fusobacteriota and Cyanobacteria. *p < 0.05
Welch’s t-test and Mann-Whitney test were performed on the phylum, family, genus and species level to compare the differences in cervicovaginal microbiota between the normal and HPV groups. At the phylum level, the Firmicutes (Fig. 2B) (p = 0.0134) was higher in the normal group than in the HPV group. The HPV group had higher numbers of the Actinobacteriota (Fig. 2C) (p = 0.0194), Fusobacteriota (Fig. 2D) (p = 0.0263), and Cyanobacteria (Fig. 2E) (p = 0.0231) were also significantly enriched compared to the normal group. Although the upregulation of fecal Firmicutes/Bacteroidetes ratio is considered an indicator of several pathological conditions [16], our results were the opposite (12.37% vs. 15.51%) in cervicovaginal condition. At the family level (Fig. 3A), Lactobacillaceae (Fig. 3B) (p = 0.0172) was more prevalent in the normal group than in the HPV group. Compared to the normal group, Bifidobacteriaceae (Fig. 3C) (p = 0.0194) Atopobiaceae (Fig. 3D) (p = 0.0466), Mycoplasmaceae (Fig. 3E) (p = 0.0014), Leptotrichiaceae (Fig. 3F) (p = 0.0254), Aerococcaceae (Fig. 3G) (p = 0.0169) and Erysipelotrichaceae (Fig. 3H) (p = 0.0462) were enriched in the HPV group.
At the genus level (Fig. 4A), compared to the normal group, the HPV group had significantly higher levels of Ureaplasma (Fig. 4B) (p = 0.0106), Aerococcus (Fig. 4C) (p = 0.0105), Sneathia (Fig. 4E) (p = 0.0253), Gardnerella (Fig. 4F) (p = 0.0050), Mycoplasma (Fig. 4G) (p = 0.0021). There were differences in Ralstonia between the two groups, but they were not statistically significant (Fig. 4D). The normal group had significantly more Lactobacillus (Fig. 4H) (p = 0.0172) than the HPV group. At the species level (Fig. 5A), Lactobacillus_iners (Fig. 5B) (p = 0.0196) in the normal group was also significantly higher than that in the HPV group. Compared to the normal group, the HPV groups had higher abundance of Veillonella_montpellie (Fig. 5C) (p = 0.0186), Ureaplasma_parvum (Fig. 5D) (p = 0.0349), Sneathia_amnii (Fig. 5E) (p = 0.0480), and Aerococcus_christensenii (Fig. 5F) (p = 0.0096).
Fig. 3
A Comparison of bacterial difference at family level. B–H the relative abundance of Lactobacillaceae, Bifidobacteriaceae, Atopobiaceae, Mycoplasmaceae, Leptotrichiaceae, Aerococcaceae and Erysipelotrichaceae. *p < 0.05, **p < 0.01
Fig. 4
A Comparison of bacterial difference at genus level. B–H the relative abundance of Ureaplasma, Aerococcus, Ralstonia, Sneathia, Gardnerella, Mycoplasma, Lactobacillus. *p < 0.05, **p < 0.01
Fig. 5
A Comparison of bacterial difference at species level. B–H the relative abundance of Lactobacillus_iners, Veillonella_montpellie, Ureaplasma_parvum, Sneathia_amnii and Aerococcus_christensenii. *p < 0.05, **p < 0.01
The LEfSe model was then used to identify specific microbiota that may be significantly associated with HPV infection (Fig. 6). The following bacteria were significantly enriched in the normal group (LDA score > 4.8): phylum of Firmicutes; family of Lactobacillaceae; genus of Lactobacillus; and species of Lactobacillus_iners. We found that the following cervicovaginal microbiota were enriched in the HPV group (LDA score > 3.6): phyla of Actinobacteriota and Zixibacteria; family of Bifidobacteriaceae, Mycoplasmataceae, and Atopobiaceae; genus of Gardnerella, Ureaplasma and Atopobium; and species of Ureaplasma_parvum.
Fig. 6
Linear discriminant analysis (LDA) integrated with effect size (LEfSe) analysis. A Cladogram indicating the phylogenetic distribution of microbiota correlated with the Normal or HPV groups. B The differences in abundance between the Normal and HPV groups
Overall overview of serum metabolites in the normal and HPV groupsA growing number of studies have found that cervicovaginal microbes are closely associated with HPV infection, and the development of cervical cancer [9, 25,26,27]. We hypothesized that changes in human metabolic pathways may be influenced by HPV. Therefore, we performed a metabolomic analysis of serum samples based on UPLC-MS untargeted metabolomics to identify potential HPV associated or microbial-related host metabolites.
We successfully identified 1488 metabolites, from both positive and negative detection mode, in the normal and HPV groups (Supplementary Table 3). A total of 32 differential metabolites were screened from the normal and HPV groups, of which 21 metabolites were down-regulated and 11 metabolites were up-regulated (Fig. 7A). Based on the relative abundance of these differentially expressed metabolites, PLS-DA was used to assess the metabolic differences between the normal and HPV groups. The results showed a significant difference in the distribution of cervicovaginal metabolites between the normal and HPV groups (Fig. 7B).
Fig. 7
Serum metabolome analysis. A Volcanogram shows differential accumulation of metabolites [log2 (fold-change) on the X-axis] and significant change [-log10 (P) on the Y-axis] between the normal and HPV groups. B Partial least squares discriminant analysis (PLS-DA) showed differences between the normal and HPV groups. C bubble map showing the enriched metabolic pathway in the normal group and HPV group
Differential metabolites were further used to predict KEGG metabolic pathways alterations (Fig. 7C and Supplementary Table 4). The identified impacted pathways included the linolenic acid metabolism, glycerol phospholipid metabolism, arachidonic acid metabolism, unsaturated fatty acid biosynthesis, lysine degradation, pyruvate metabolism, primary cholecystic acid biosynthesis. Pathway enrichment analysis based on the relative abundance of metabolites showed that the KEGG metabolic pathway of α-linolenic acid (ALA) metabolism was significantly enriched (Fig. 7C) and two metabolites (Phosphatidylcholine, (9Z,12Z,15Z)-Octadecatrienoic acid) were directly involved. Enrichment results for specific pathways are shown in Supplementary Table 5. The main enrichment pathway was related to linolenic acid metabolism. Heat map analysis showed significant differences in metabolic patterns between the normal and HPV groups based on the identified biomarkers and metabolites of the enriched pathways (Fig. 8).
Fig. 8
Heat maps of identified key metabolites for differentiation of normal and HPV groups
Identification of specific metabolites for HPV infectionTo further identify metabolite changes associated with HPV infection, biomarker analysis was performed. The area under the ROC curve for the 13 variable model was 0.823 (CI: 0.689–0.94) (Fig. 9A). The predicted probability of classification for all samples indicated that the normal and HPV groups were well classified (Fig. 9B). Figure 9C shows the 13 most important biomarkers, including 9,12,13-Trihydroxy-10(E),15(Z)-octadecadienoic acid, PC (18:0/22:6), PC (18:3 (6Z,9Z,12Z) /18:1(9Z)), α-linolenic acid, 9,10-DiHOME, PC (18:3 (6Z,9Z,12Z) /P-18:1 (11Z)), pipecolic acid, O-cresol, prostaglandin F3α, ethylparaben, S-lactoylglutathione, glycocholic acid, and 3-methylcrotonylglycine. The ROC curves of these potential candidate biomarkers showed that the AUC of all candidate biomarkers was above 0.656 with a P value less than 0.05, indicating that they might be significantly associated with HPV infection (Supplementary Table 6).
Fig. 9
Important discriminatory metabolites identified by correlation and multivariate analysis between the Normal and HPV groups. A ROC curve based on cross validation (CV) performance. The variables used in the model are displayed in C below. B the predictive class probabilities for each sample based on AUC. C The PLS-DA model obtained significant discriminatory metabolites of average importance
Correlation analysis reveals an overview of the microbial metabolic axis of HPV infectionBased on the microbiome and metabolomics data described above, we subsequently performed Pearson correlation analysis to identify relevant microorganisms and metabolites in the HPV and normal groups.
As shown in Fig. 10, 9,10-diHOME was significantly positively correlated with Sneathia (p = 0.0006). The ALA was positively correlated with Sneathia-amnii (p = 0.0412) and Sneathia (p = 0.0008), but was significantly negatively correlated with Lactobacillus and Lactobacillus_iners. Ethylparaben was positively correlated with Atopobium (p = 0.0077), Sneathia (p = 0.0077) and Mycoplasma (p = 0.0069). Glycocholic acid (p = 0.0069) was positively correlated with Atopobium (p = 0.00002) and Ralstonia (p = 0.00001). Pipecolic acid was negatively correlated with Sneathia (p = 0.0161) and Mycoplasma (p = 0.0026). Gardnerella was positively correlated with 9,12,13-Trihydroxy-10(E),15(Z)-octadecadienoic acid (p = 0.0492). Lactobacillus_iners was positively correlated with PC (18: 3(6Z,9Z,12Z) / 18: 1 (9Z)) (p = 0.0120). The results showed that 9,10-DiHOME, ALA, ethylparaben, glycocholic acid, pipecolic acid, 9,12,13-trihydroxy-10(E),15(Z)-octadecadienoic acid, PC (18:3(6Z,9Z, 12Z) / 18:1(9Z)) and PC (18:3(6Z,9Z,12Z) / P-18: 1 (11Z)), correlating with Sneathia (Sneathia-amnii), Lactobacillus (Lactobacillus_iners), Atopobium, Mycoplasma, and Gardnerella, may be potential biomarkers of HPV infection.
Fig. 10
Integrated correlation-based analysis (Pearson’s correlation) of key altered microbes and metabolites upon HPV infection
留言 (0)