
記住我









Myocardial fibrosis is a common pathophysiologic companion of many different myocardial conditions, where the cardiac interstitium expands through the deposition of extracellular matrix (ECM) proteins (Frangogiannis, 2021). Unlike other organs, the adult mammalian heart has limited regenerative potential. In response to ischemic insults, systemic diseases, or any other harmful stimulus to the circulatory system or the heart itself, the damaged cardiomyocytes are replaced by a fibrotic scar (Gyöngyösi et al., 2017). Though this event is crucial for the preservation of ventricular rupture (Travers et al., 2016), over time, excessive and continuous ECM deposition leads to irreversible ventricular remodeling and distorted organ geometry and significantly impairs the function of the heart (Li et al., 2018; Maruyama and Imanaka-Yoshida, 2022; Majid et al., 2023).
A wide repertoire of cell populations, including cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and pericytes; different types of immune cells (myeloid and lymphoid); adipocytes; mesothelial cells; and neuronal cells synchronously maintain cardiac function (Meilhac and Buckingham, 2018; Litviňuková et al., 2020; Tucker et al., 2020; Marín-Sedeño et al., 2021). Sustenance of cardiac homeostasis depends on the integrity of individual cells and the cellular interactions mediated by juxtacrine, paracrine, and endocrine signals. Any pathogenic stimuli disrupting the cardiac microenvironment, metabolic demand, hemodynamic stability, or these crosstalk networks strain the cardiomyocytes and lead to counter-responses (Pogontke et al., 2019; Marín-Sedeño et al., 2021; Liu et al., 2023) and initiation of an inflammatory cascade (Hara and Tallquist, 2023). Activation of the resident immune cells as well as recruitment of innate and adaptive immune cells attempt to adapt to the insult. However, the activation of pattern recognition receptors (PRRs) by damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) initiates downstream signaling cascades that might upregulate the expression of genes encoding pro-inflammatory cytokines and chemokines (Mann, 2011; Adamo et al., 2020a; Silvis et al., 2020). Persistent low-grade inflammation activates tissue-resident macrophages and mast cells to recruit and activate B and T cells, which trigger plasma protein infiltration. The temporal imbalance between the activity of matrix metalloproteinases (MMPs) and tissue inhibitor of MMPs (TIMPs) with a collateral increase in transforming growth factor-β (TGF-β) signaling and NLR family pyrin domain-containing 3 (NLRP3) inflammasome formation leads to aberrant fibrotic deposition and left ventricular (LV) remodeling (Mezzaroma et al., 2011; Zhang et al., 2011; Adamo et al., 2020a). Changes in cardiac performances resulting from altered intercellular signaling, loss of cell activity, or cell death can then result in further changes in cell-to-cell communication (Buckingham et al., 2005; Marín-Sedeño et al., 2021) (Figure 1).
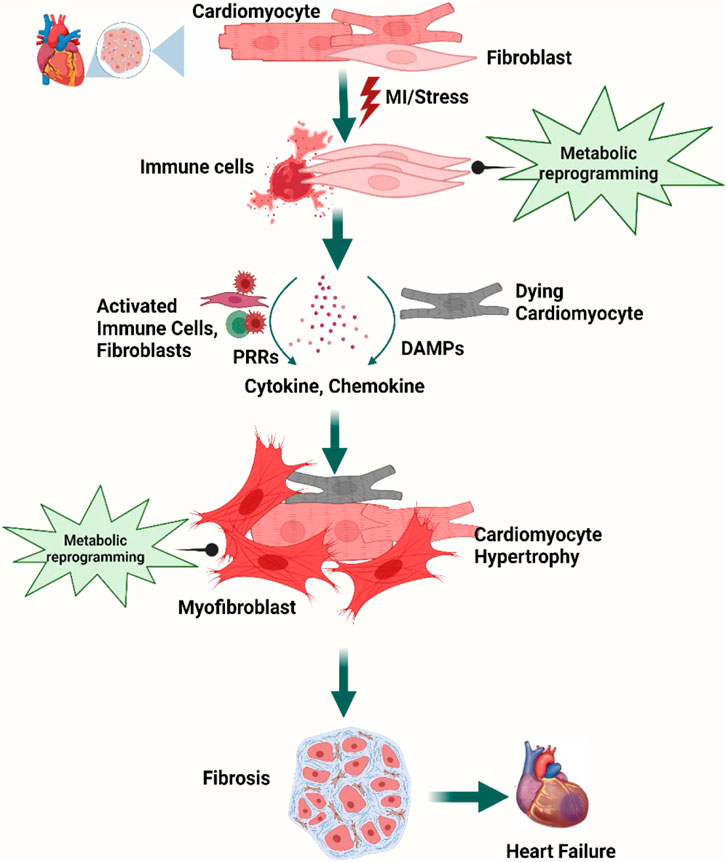
Figure 1. Overview of cellular steps leading to cardiac fibrosis. Following injury, cardiac fibroblasts are activated and transdifferentiated into myofibroblasts to replace dying cardiomyocytes. Dying myocytes release damage-associated molecular patterns (DAMPs) which activate immune cells and trigger downstream signaling cascades of pro-inflammatory cytokines and chemokines. A plethora of cytokines and chemokines in addition to myofibroblasts undergo metabolic alterations that drive the development and progression of myocardial fibrosis and heart failure. Figure created using BioRender.
Along with immune activation, metabolic reprogramming, supply–demand mismatch, and disruption of the equilibrium in local metabolites in the affected cardiac tissue contribute to progression of cardiac fibrosis. Metabolic reprogramming allows the cells to adopt dynamic changes in cellular metabolic pathways and biological functions in response to various stimuli or environmental conditions, for example, cellular metabolism switches from oxidative metabolism (i.e., oxidative phosphorylation, OXPHOS) to the more oxygen-sparing carbohydrate metabolism (i.e., glycolysis) and utilization of glutamine and fatty acid increases to meet the energy and biosynthetic demands during acute and chronic cardiac stress (Sun et al., 2020a; Sabogal-Guáqueta et al., 2023; Ritterhoff and Tian, 2017; Rosano and Vitale, 2018; Yoganathan et al., 2023; Razeghi et al., 2001; Akki et al., 2008; Aubert et al., 2016; Bedi et al., 2016; Tran and Wang, 2019; Lin et al., 2023). With progressive low-grade inflammation in the background of cardiac insult, cytokines and nutrient metabolites activate inflammatory programs through shared pathways, and resident and recruited immune cells undergo metabolic shifts on their own as well during fibrotic events (Sun et al., 2020a; Sabogal-Guáqueta et al., 2023). The energy demand and metabolic intermediates for immune cell activation and differentiation are dependent more on glycolysis than on the tricarboxylic acid (TCA) cycle and OXPHOS (Srivastava et al., 2018; Cho et al., 2020; Farah et al., 2021; Wenzl et al., 2021). Moreover, the fibroblasts adopt a metabolic phenotype using glycolysis and glutamine-derived α-ketoglutarate to adapt to the altered microenvironment (Kawaguchi et al., 2011; Mouton and Hall, 2020; Farah et al., 2021; Francisco et al., 2021).
Immunometabolism is still a burgeoning field, and much of the existing knowledge on cardiac immune cell metabolism is based on myocardial infarction (MI) models (Mouton and Hall, 2020). In the past few years, technological advances and highly sensitive metabolomics approaches have redefined the inextricable relationships between immune activation, molecular signaling, and metabolism and discovered their association to immune cell functions in the course of the disease. However, there are knowledge gaps in the complete sketch of immunometabolism from the perspective of cardiac fibrosis that require further exploration to use immune modulation strategies to prevent cardiac remodeling.
In this review, we provide an update on the current understanding of the involvement of immune and metabolic systems in cardiac fibrosis and the potential of immune–metabolic reprogramming in the management of cardiac fibrosis and restoration of cardiac function.
2 Contribution of the immune system to cardiac homeostasisA large number of innate and adaptive immune cells are found in the heart. Following infiltration of the cardiac tissue at gestation, the immune cells persist in the myocardium and engage in essential housekeeping functions; defend against pathogens, toxic insults, hypoxia, or other injury; and maintain normal cardiac function throughout life. Though the resident and recruited immune cell populations change in different stages of life as well as over the course of injurious stimuli, the major subtypes of inflammatory cells include leukocytes, mononuclear phagocytes, neutrophils, B cells, and T cells. The complex interactions and crosstalk between different subsets of immune cells that either reside or infiltrate the cardiac tissue and the resident cardiac and non-cardiac cells comprising cardiomyocytes, fibroblasts, and endothelial cells maintain the physiological microenvironment in the heart (Pinto et al., 2012; Ramos et al., 2017; Swirski and Nahrendorf, 2018; Marelli-Berg and Aksentijevic, 2019; Steffens et al., 2022).
Architecturally, the heart is heterogeneous, as is the distribution of immune cells: various macrophage subsets are non-uniformly distributed in distinct niches of the heart. Dendritic cells are found abundantly in the cardiac valves and aortic sinus (Choi et al., 2009), whereas the atrioventricular node contains a high concentration of tissue-resident macrophages (Hulsmans et al., 2017). On the other hand, the coronary vasculature is rich in CCR2- (CC chemokine receptor 2) macrophages and fetal monocyte-derived macrophages are concentrated adjacent to the endocardial trabeculae (Leid et al., 2016). These findings suggest that biochemical, neurohormonal, nutritional, or metabolic alterations of different niches of the heart involve different populations of inflammatory cells as principal responders and lead to different pathophysiological courses. Moreover, the organized chambers, vessels, and myocardium are immersed in the serosal fluid within the pericardium, which contains leukocytes, macrophages, and B cells and provides tissue-infiltrating leukocytes during challenges (Butts et al., 2017). The pericardial adipose tissue supplies lymphocytes and coordinates granulopoiesis and the activation of immune cells (Horckmans et al., 2018), whereas white adipose tissue synchronizes the supply and mast cell accumulation in the heart following MI (Ngkelo et al., 2016). We lack a complete picture of the spatiotemporal distribution of different immune cell populations. A summary of the immune cells along with their distribution and role in the heart is enlisted in Table 1.
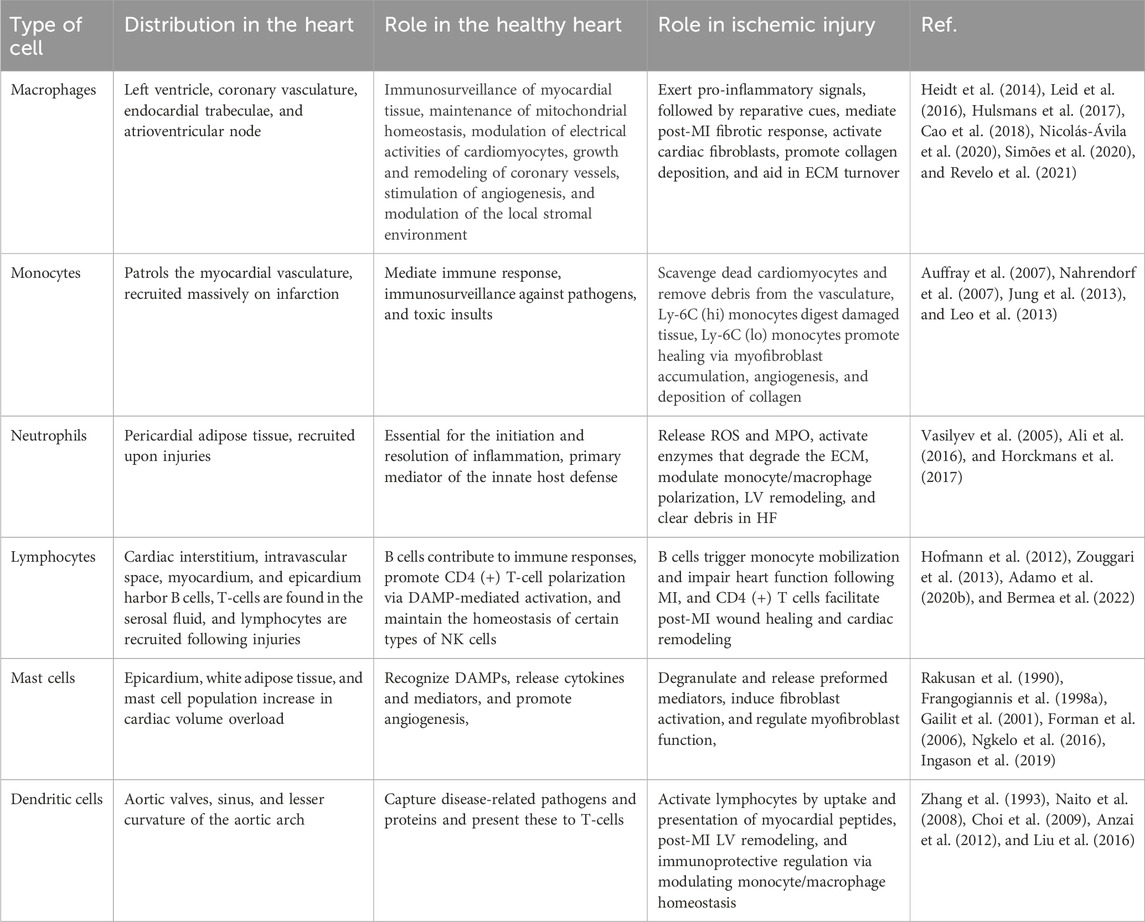
Table 1. Role of different immune cells in healthy and ischemic heart.
Specific localization of different immune cells in the heart suggests that they have specific interactions with resident cardiac cells. Cardiomyocytes, fibroblasts, and endothelial cells not only express receptors that recognize inflammatory mediators from inflammatory cells but also produce growth factors, cytokines, and chemokines to which leukocytes respond. Mast cell-derived tumor necrosis factor (TNF) activates endothelial cells (Frangogiannis et al., 1998a), IL-6 from cardiomyocytes activates neutrophils via intercellular adhesion molecule 1 (ICAM1) expression (Gwechenberger et al., 1999), IL-17 from T cells stimulates cardiac fibroblasts (Wu et al., 2014), and fibroblast-derived granulocyte–macrophage colony-stimulating factor (GM-CSF) induces the production and recruitment of myeloid cells (Anzai et al., 2017). On the other hand, macrophage-derived TGFβ, vascular endothelial growth factor (VEGF), and IL-10 promote collagen production, neo-angiogenesis, and resolution of inflammation (Nahrendorf et al., 2007; Hilgendorf et al., 2014) (Figure 2).
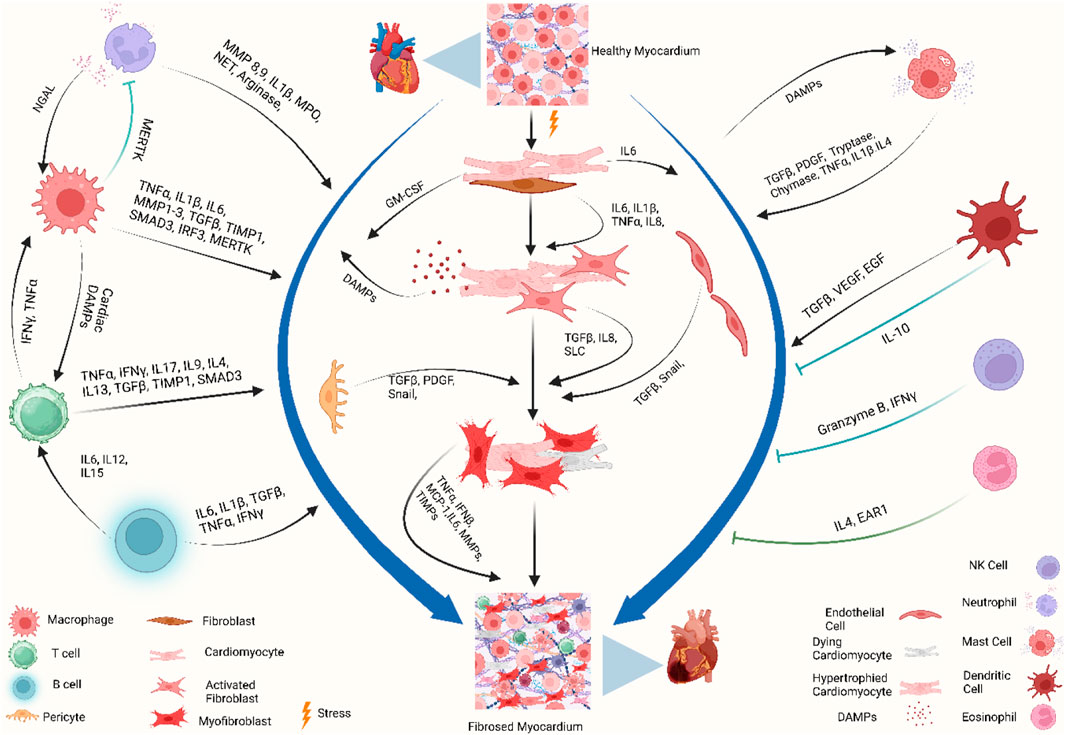
Figure 2. Immune crosstalk between inflammatory cells and cardiac cells during the transformation of the cardiac microenvironment toward fibrosis. The injured myocardium alters the cardiac microenvironment and releases different growth factors and inflammatory mediators, i.e., IL-6, IL-1β, GM-CSF, TNF-α, PDGF, and DAMPs, from injured cardiomyocytes, cardiac fibroblasts, endothelial cells, and pericytes (Gwechenberger et al., 1999; MeléNdez et al., 2010a; Anzai et al., 2017). Upon activation, cardiac fibroblasts further promote the cascades and release MMPs, TIMPs, MCP-1, and TNF-α, which play a role in ECM remodeling and fibrogenesis (Yoshimura et al., 2014; Pluijmert et al., 2021). During these events, the recruited and activated immune cells release another wave of cytokines and chemokines which further promote cardiac remodeling. Recruited monocytes and macrophages trigger inflammatory responses by releasing DAMPs, detect extracellular DNA from dying cells, and release type I interferons (King et al., 2017). Resident macrophages, IL-10 signaling, and MERTK modulate inflammatory cells, promote reparative events, inhibit hyperactivation of fibroblasts, and mitigate fibrosis (DeBerge et al., 2017; Jung et al., 2017; Revelo et al., 2021). Simultaneously, neutrophils, lymphocytes, and mast cells crosstalk with macrophages and fibroblasts to stage the background of ECM turnover and deposition of collagen by releasing inflammatory cytokines. Dendritic cells and NK cells contribute to improving fibrosis by modulating adverse inflammatory events (Ong et al., 2015; Choo et al., 2017). When these profibrotic interplay and crosstalk between different resident and non-resident immune and non-immune cells in the stressed heart outweigh the reparatory mechanisms, the consequential events ultimately lead to cardiac fibrosis. Abbreviations: MERTK, proto-oncogene tyrosine-protein kinase MER; NGAL, neutrophil gelatinase-associated lipocalin; IFNγ, interferon gamma; TNF-α, tumor necrosis factor-alpha; IL1β, interleukin-1 beta; IL6, interleukin 6; IL15, interleukin 15; IL12, interleukin 12; IL8, interleukin 8; IL17, interleukin 17; IL9, interleukin 9; IL4, interleukin 4; IL13, interleukin 13; MMP, matrix metalloproteinase; TIMP, tissue inhibitors of metalloproteinases; SMAD, suppressor of mothers against decapentaplegic; IRF3, interferon regulatory factor 3; MPO, myeloperoxidase; NET, neutrophil extracellular traps; GM-CSF, granulocyte–macrophage colony-stimulating factor; DAMP, damage-associated molecular pattern; Snail, zinc finger protein SNAIL1; SLC, solute carrier; MCP-1, monocyte chemoattractant protein-1; VEGF, vascular endothelial growth factor; EGF, epidermal growth factor; EAR1, eosinophil-associated ribonuclease 1. Figure created using BioRender.
Newer subsets of immune cells and novel roles of different inflammatory cells are being revealed with ongoing research. Leukocytes and their products are gaining focus in the context of normal physiological as well as pathological fibrotic events. A brief discussion of the major contributors to cardiac fibrosis is provided in the following section.
2.1 Monocytes and macrophages in cardiac fibrosisCardiac macrophages are part of a steady-state cell network that contributes not only to forming a repertoire of immune cells but also to maintaining the mechanically strenuous and energy-intense pumping function of the heart. They electrically couple to cardiomyocytes through connexin 43 (CX43, also known as GJA1)-containing gap junctions in normal mouse and human hearts (Fernández-Ruiz, 2017). Macrophage ablation results in cardiac pathologies such as progressive atrioventricular block and conduction abnormalities in the atria and ventricles (Hulsmans et al., 2017). Moreover, macrophages help in cell and matrix turnover and regulate angiogenesis and matrix deposition and removal in cardiac pathologies (Guo et al., 2018; Simões et al., 2020; Li et al., 2023a). Hence, spatiotemporal alteration and phenoconversion of macrophage populations in cardiac fibrosis not only remodel the structure but also cardiac rhythmicity (Figure 2).
2.1.1 Subsets of cardiac macrophagesMacrophages and monocytes regulate fibrotic responses across many tissues (Wynn and Ramalingam, 2012). The myocardium of adult mammals typically has a restricted number of resident macrophages (Gersch et al., 2002; Heidt et al., 2014; Mylonas et al., 2015) which play a role in maintaining cardiac homeostasis (Hulsmans et al., 2017). Moreover, the development of sophisticated tools, such as single-cell RNA sequencing, has found evidence of heterogeneous and ontogenetically diverse macrophages in the heart. At least seven subsets of cardiac and pericardial macrophage populations have been identified in the infarcted heart with unique spatiotemporal dynamics and morphologic characteristics (Bajpai et al., 2018; Wei et al., 2023). Tissue-resident cardiac macrophages are principally divided based on the expression of CC-chemokine receptor 2 (CCR2). CCR2- macrophages are of embryonic origin, and they seed the cardiac tissue during embryonic and early postnatal development. They support the development of coronary vasculature (Leid et al., 2016), cardiac regeneration, and electrical conduction within the atrioventricular (AV) node (Aurora et al., 2014; Lavine et al., 2014; Hulsmans et al., 2017). On the other hand, adult hematopoietic lineages produce CCR2+ macrophages, which facilitate neutrophil extravasation and monocyte recruitment (Li et al., 2016) and initiation of inflammation within the diseased heart (Epelman et al., 2014b; Heidt et al., 2014; Epelman et al., 2015; Bajpai et al., 2019). Another recent study reported the existence of a GATA6+ population of macrophages with an anti-fibrotic role within the pericardial cavity (Deniset et al., 2019). The dynamics of macrophage lineages change over time depending on the injurious stimuli and try to adopt changes to cope with the altered microenvironment. Thus, different subsets of macrophages contribute to the maintenance as well as pathological events of the heart.
2.1.2 Macrophage plays an integral role in cardiac fibrosisThe macrophage heterogeneity influences the outcome of myocardial injuries in neonatal and adult hearts. The neonatal heart contains only CCR2- cardiac resident macrophages, while the adult heart contains both CCR2- and CCR2+ macrophage populations (Aurora et al., 2014; Lavine et al., 2014). Following an injury, for example, after an MI, the neonatal heart expands CCR2- macrophages, while the adult heart expands CCR2+ macrophages. In the adult heart, monocyte-derived CCR2+ macrophages are recruited by the inflammatory signal transduction pathways, DAMPs, and other chemokine-driven pathways and replace the CCR2- population (Dewald et al., 2005; Frangogiannis et al., 2007). Upon recruitment, the CCR2+ population continues the phagocytic activities and promotes inflammation by activating the inflammatory bodies, by pattern recognition receptor (PRR) signaling, and by producing cytokines (Epelman et al., 2014a; Heidt et al., 2014; Sager et al., 2016; Bajpai et al., 2019; Chen et al., 2023; Isidoro and Deniset, 2023). These macrophages play a role in regulating inflammation, promoting fibrosis, facilitating matrix remodeling, supporting angiogenesis, and contributing to the process of regeneration. They release different cytokines, such as interleukin-1β, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-16 (IL-16), interleukin-18 (IL-18), transforming growth factor-β (TGF-β1), platelet-derived growth factor (PDGF), matrix metalloprotease-9 (MMP-9), and tissue inhibitor of metalloproteinase 1 (TIMP 1), and induce alpha-smooth muscle actin (αSMA), lysyl oxidase (LOX), and collagen type 1 alpha 2 (Col1a2) expression in fibroblasts, thus exhibiting profibrotic activity (Lindsey et al., 2006; MeléNdez et al., 2010a; Tamaki et al., 2013; de Couto et al., 2015; Frangogiannis, 2015; Chen and Frangogiannis, 2017; Honold and Nahrendorf, 2018; Liao et al., 2020; Moskalik et al., 2022). On the other hand, the macrophages with a reparatory phenotype (CCR2-) release IL-10 after the acute phase and attempt to repair cardiac function (Krishnamurthy et al., 2009; Jung et al., 2017; Moskalik et al., 2022). Metabolic reprogramming further polarizes macrophages into pro-inflammatory (M1) or anti-inflammatory (M2) type and, in turn, this phenoconversion orchestrates the background of scarring or healing of the injured myocardium (Kim et al., 2021; Kang et al., 2023) (Figure 2). Moreover, subsets of myeloid cells can engage in the fibrosis process by being converted to fibroblast-like cells or differentiating fibroblasts into myofibroblasts (Meng et al., 2016; Sinha et al., 2018; Haider et al., 2019; Simões and Riley, 2022; Wu et al., 2022).
2.2 Granulocytes in cardiac fibrosis2.2.1 Direct role of neutrophils in cardiac remodelingThe role of neutrophils in the regulation of fibrosis is context-dependent. Reperfusion of the post-ischemic myocardium promotes neutrophil infiltration, which exacerbates the pro-inflammatory response and contributes to the ischemia–reperfusion (I/R) injury of the ischemic border zone. They produce and release reactive oxygen species (ROS) and myeloperoxidase (MPO), resulting in the generation of cytotoxic aldehydes, oxidative stress, activation of enzymes degrading the ECM and causing cardiomyocyte apoptosis, and maladaptive remodeling (Vinten-Johansen, 2004; Vasilyev et al., 2005; Fröhlich et al., 2013; Koeth et al., 2013; Puhl and Steffens, 2019; Okyere and Tilley, 2020). However, neutrophils exhibit a strong association with acute inflammatory reactions in the myocardium and play a role in tissue repair by influencing the behavior of macrophages (Horckmans et al., 2017). In a mouse model, neutrophils have been shown to direct macrophages toward a reparative phenotype through the secretion of neutrophil gelatinase-associated lipocalin (NGAL) which mediates efficient clearance of debris. Moreover, antibody-mediated depletion of neutrophils increases cardiac fibrosis and worsens cardiac function (Horckmans et al., 2017) (Figure 2). Therefore, in regulation of fibrosis and for favorable cardiac remodeling, a balanced neutrophil response plays a crucial role.
Studies have found a high plasticity potential of neutrophils and their roles in modulating the outcome of inflammatory events. Ma et al. (2016) reported that the temporal polarization of neutrophils toward pro-inflammatory N1 exacerbates left ventricle (LV) remodeling, whereas the anti-inflammatory N2 phenotype attenuates adverse LV remodeling. DAMPs released from necrosed myocytes stimulate N1 polarization by stimulating TLR4, whereas TGF-β1 inhibition attenuates pro-inflammatory neutrophils (Ma et al., 2016).
The presence of neutrophils in the wounded region is transient since they are rapidly eliminated. Replacement of neutrophils with Ly6Clow macrophages is aligned with the transition from the inflammatory phase to the reparatory phase and decreased production of inflammatory cytokines, growth factors, and chemokines (Nahrendorf et al., 2007). Therefore, the involvement of neutrophils in chronic cardiac fibrosis is restricted to the initial phases of fibroblast activation. According to a recent study, a noteworthy mechanism depends on neutrophils and can potentially contribute to age-related cardiac fibrosis. Within the core of an aging organism, the stimulation of ROS in neutrophils can initiate the creation of neutrophil extracellular traps (NETs). This process is facilitated by activation of the peptidyl arginine deiminase 4 enzyme (PAD4). In vivo, tests indicate that the production of NETs through the involvement of PAD4 plays a role in the development of interstitial fibrotic alterations and the onset of left ventricular diastolic dysfunction (Martinod et al., 2017) (Figure 2).
2.2.2 Contribution of basophils to cardiac fibrosisBecause of their rarity, basophils have long been overlooked in immunological research. Basophils circulate in the bloodstream under homeostatic conditions, but they infiltrate tissues during inflammation (Miyake et al., 2022). Despite their low numbers, basophils affect the accumulation of myeloid cells and influence cardiac remodeling. Sicklinger et al. (2021) showed that basophil depletion promoted a shift from Ly6Clow macrophages with reparatory phenotype toward inflammatory Ly6Chi monocytes in the infarcted myocardium. Induction of IL-4 and IL-13 by glycoprotein IPSE/α-1 in basophils improves cardiac functions and post-MI cardiac healing (Sicklinger et al., 2021). The role of basophils in cardiac fibrosis is further corroborated by the finding that low blood basophil counts are associated with increased scar size and poor outcomes in patients with acute MI (Miyake et al., 2022). Moreover, IL-4 released from the infiltrating basophils acts on resident fibroblasts, triggers myofibroblast expansion, and leads to the production of connective tissue elements from myofibroblasts (Schiechl et al., 2016).
2.3 Lymphocytes in cardiac fibrosis2.3.1 B cells in cardiac fibrosisThe adaptive immune cells, B and T lymphocytes, are found in small numbers in a normal physiological heart but increase following injury (Zouggari et al., 2013; Wang et al., 2019). The B-cell population in the human heart is divided between the intravascular space and the interstitial space (Bermea et al., 2022) and further divided into subgroups. B1 and B2 cells contribute to the innate immune response through secretion of IFN-γ, IL-6, and IL-17; promote CD4+ T-cell polarization; and regulate the mobilization of monocytes through the production of CCL7, whereas Bregs or B10 cells support immunologic tolerance, resolve the acute inflammatory response, and maintain the homeostasis of certain types of natural killer cells through the secretion of IL-10, IL-35, and TGF-β (Zouggari et al., 2013; Chimen et al., 2015; Shen and Fillatreau, 2015) (Figure 2).
Cardiac B cells in neonatal mice promote cardiomyocyte proliferation, angiogenesis, and regeneration of the heart and inhibit inflammatory responses, while adult B cells promote inflammation and impair cardiac function following myocardial injury (Zouggari et al., 2013; Tan et al., 2023). Depletion of neonatal B cells reduces cardiac regeneration and promotes fibrotic scarring in the post-MI heart, whereas B-cell depletion in adult mice inhibits myocardial fibrosis and improves cardiac function (Tan et al., 2023). Moreover, activated B cells contribute to sustained immune system activation and myocardial inflammation, promote the synthesis of myocardial collagen types I and III, and damage the left ventricular ejection fraction (Mo et al., 2021). Studies have found that B cells promote fibrosis through releasing inflammatory cytokines like IL-1β, IL-6, and TNFα, whereas depletion of B cells results in attenuation of collagen deposition following MI, transverse aortic constriction, and nonischemic cardiomyopathy (Yu et al., 2013; Cordero-Reyes et al., 2016; Ma et al., 2018).
2.3.2 Heterogeneity of T-cell populations in cardiac fibrosisT-cell receptor engagement, antigenic stimuli, tissue microenvironment, and metabolic reprogramming shape the repertoire of T cells into that of T helper cells (Th1, Th2, Th9, Th17, and Th22), cytotoxic T lymphocytes (CTLs), regulatory T (Treg) cells, and natural killer T (NKT) cells (Zhang and Zhang, 2020). Induction of T cells by cardiac DAMPs processed by antigen-presenting cells results in cardiotropism, transformation of cardiac fibroblasts, and maladaptive cardiac remodeling (Bayer et al., 2023) (Figure 2). An increasing body of literature indicates that distinct subpopulations of T lymphocytes play a substantial role in the direct stimulation of fibroblasts and progression of cardiac fibrosis (Nevers et al., 2015; Li et al., 2017; Abdullah and Jin, 2018). One study proposed the presence of a binding interaction between activated Th1 cells and cardiac fibroblasts in the left ventricular pressure overload model. The interaction has the potential to trigger the synthesis of TGF-β by fibroblasts, resulting in the differentiation of fibroblasts into myofibroblasts (Nevers et al., 2017). Furthermore, an increased number of Th2 cells in cardiac tissues are afflicted with fibrosis (Duerrschmid et al., 2015). The behavior under observation can be attributed to the increased regulation of profibrotic cytokines, such as IL-4 and IL-13, which effectively enhance collagen synthesis by fibroblasts. The infiltration of Th17 cells into the fibrous myocardium has been suggested to be involved in the pathogenesis of autoimmune myocarditis, contributing to the progression of a fibrous myocardium (Baldeviano et al., 2010). Moreover, T cells can stimulate fibroblast activation via their fibrogenic activity. The profibrotic impact produced by T lymphocytes suggests their involvement in the survival of cardiac elastocytes, resulting in the replacement of dead cells by fibrous tissues (Kallikourdis et al., 2017).
2.3.3 Emerging insights regarding the role of T cells in cardiac fibrosisAlthough the precise influence of different T-cell subpopulations on the development of fibrosis remains unclear, a growing body of evidence indicates that the use of regulatory T cells (Tregs) in cellular therapy holds promise in reduction of the incidence of myocardial infarction and consequently the ensuing fibrotic response (Kvakan et al., 2009; Tang et al., 2012). Tregs could reduce the fibrogenic activity of macrophages and CD8+ T-cell depletion after experimental MI in mouse models, which reduces inflammation and preserves ventricular function (Weirather et al., 2014; Santos-Zas et al., 2021). Furthermore, Saxena et al. (2014) have reported that Tregs could impact the phenotype of fibroblasts. Moreover, they have been observed to secrete signals that inhibit fibrosis (Tang et al., 2012); however, the precise characteristics of these signals have yet to be elucidated. Notably, Tregs can secrete fibrous mediators, such as TGF-β (Taylor et al., 2006; Saxena et al., 2014; Wang et al., 2023). Hence, the regulatory role of these cells in the fibrotic response is likely dependent on the specific circumstances and the balance between fibrotic and anti-fibrotic cellular processes.
2.3.4 Emerging role of NK cells in cardiac fibrosisNatural killer cells (NK cells) are type-I innate lymphoid cells known for their role in the recognition and elimination of virus-infected and malignant cells and in limiting their spread (Chiossone et al., 2018). NK cells also modulate immune responses by reciprocally interacting with macrophages, dendritic cells, T cells, and endothelial cells and contribute to the regulation of cardiac diseases (Vivier et al., 2008; Ong et al., 2017). Sustained NK cell deficit in patients with coronary artery disease was associated with persistent low-grade inflammation in the heart (Backteman et al., 2013). As an unabated chronic inflammation plays a role in eventual fibrotic remodeling of the cardiac tissue, NK cells might play a potential role in repairing and maintaining tissue homeostasis by modulating the inflammatory cascades (Tosello-Trampont et al., 2017). Activated NK cells aggregate in the heart, release granzyme B and IFN-γ, and exhibit increased expression of activation markers such as CD69, TRAIL (tumor necrosis factor-related apoptosis-inducing ligand treatment), and CD27. Hyperactivation of NK cells suppresses eosinophil activation and causes eosinophil apoptosis in an anti-inflammatory milieu, leading to decreased cardiac fibrosis (Ong et al., 2015). Moreover, NK cells lower the expression of eosinophil-related chemokines, eotaxin 1 (CCL11), eotaxin 2 (CCL24), CXCL9, and CXCL10 by resident cardiac fibroblasts (Ong et al., 2015). In post-myocardial injuries, NK cells lower cardiomyocyte apoptosis, collagen deposition, and consequent fibrosis and promote neovascularization (Ayach et al., 2006; Boukouaci et al., 2014). However, the role of NK cells in MI still lacks full characterization, and more research studies are required to explore the potential of NK cells in limiting the deposition of collagen and development of cardiac fibrosis.
2.4 Mast cells in cardiac fibrosis2.4.1 Mast cells do more than allergic reactionThe myocardium of adult animals harbors a limited population of mast cells. Notably, the abundance of mast cells in big mammals, such as dogs, surpasses that observed in mice (Frangogiannis et al., 1999). Cardiac mast cells are multifaceted resident immune sentinel cells playing a pivotal role in fibrotic remodeling in response to various myocardial injuries (Legere et al., 2020; Jin et al., 2022). Heart failure has been found to correlate with elevated quantities of mast cells (Shiota et al., 2003; Wei et al., 2003; Somasundaram et al., 2005; Luitel et al., 2017; Guimbal et al., 2021). Mast cells recognize DAMPs through TLR and ST2 (interleukin 1 receptor-like 1 receptor) and release preformed mediators and synthesize and secrete cytokines, chemokines, and lipid mediators (Janicki et al., 2015; Shao et al., 2015; Jin et al., 2022). The precise mechanisms responsible for increasing mast cells in the fibrotic cardiac area have yet to be fully elucidated. The growth factor known as stem cell factor (SCF) is crucial in the recruitment, development, and proliferation of fully developed mast cells. Additionally, it has been suggested that SCF may contribute to the localized increase in mast cells in the heart, thereby influencing cardiac pathology (Frangogiannis et al., 1998a). The origin of mast cells that infiltrate the diseased myocardium could be derived from adipose tissue (Ngkelo et al., 2016). Though the role of mast cells in the angiogenic responses in hypoxic tissues and cardiac fibrosis is reported, it lacks a complete picture. The pathophysiological role of mast cells depends on the tissue microenvironment and can be both pro- or anti-fibrotic in nature (Legere et al., 2019) (Figure 2).
2.4.2 Mast cells contribute to experimental cardiac fibrosisExperimental data show that mast cell growth significantly impacts cardiac fibrosis progression (Levick et al., 2011; Levick and Widiapradja, 2018; Widiapradja et al., 2019). In a mouse model of left ventricular pressure overload, mice without mast cells showed decreased perivascular fibrosis, which is associated with decreased progression to decompensated heart failure (Hara et al., 2002). After pressure overload, mast cells accumulate in the artery, contributing to ventricular fibrillation through platelet-derived growth factor-A (PDGF-A) expression (Liao et al., 2010). In hypertensive rat models, the stabilization of mast cells reduces fibrotic cardiac remodeling by preventing myocardial infiltration by macrophages (Levick et al., 2009). On the other hand, cardiac fibroblasts showed a profibrotic phenotype in response to mast cell mediators in mice with cardiac-specific overexpression of TNF (tumor necrosis factor) (Zhang et al., 2011). The significant profibrotic effects of mediators derived from mast cells are supported by the findings that left ventricular diastolic dysfunctions are present in many patients with systemic activation disorders of mast cells (Kolck et al., 2007).
2.4.3 Mast cell derivatives activate fibroblasts and promote fibrosisMast cells can store a diverse array of preformed fibrogenic mediators within granules alongside their capacity to generate cytokines and growth factors (Grützkau et al., 1998; Patella et al., 1998). These synthetically generated bioactive chemicals exhibit notable effectiveness in stimulating fibroblasts upon exposure to external stimuli. Following an injury, mast cell degranulation can be initiated through a range of mechanisms, such as the activation of the complement system, the formation of ROS, the activation of adenosine receptors, or the stimulation of cytokines (Frangogiannis et al., 1998a; Murray et al., 2004; Meléndez et al., 2010b). The process of degranulation results in the release of substantial quantities of fibrous agents, which have the potential to initiate or intensify the fibrous response, such as TNF-α (Frangogiannis et al., 1998a), TGF-β (Shiota et al., 2003), IL-4 (Kanellakis et al., 2012), and PDGF (Nazari et al., 2016). Nevertheless, it is important to acknowledge that fibrinogen production is not limited solely to mast cells. Various additional cell types, such as macrophages, lymphocytes, vascular cells, and myocytes, collectively contribute to the pathogenesis of cardiac fibrosis, as discussed earlier. The precise involvement of mast cells in this pathway has yet to be determined, although histamines, tryptases, and chymases can substantially influence the fibrotic process due to their distinct localization within mast cells (de Almeida et al., 2002) (Figure 2).
Chymases can generate angiotensin II (Urata et al., 1990), potentially making it a key mast cell-derived mediator in cardiac fibrosis. It has been proposed that over 75% of cardiac-specific angiotensin II in failing hearts may come from the chymase pathway, independent of ACE (angiotensin-converting enzyme) (Urata et al., 1990). This pathway remains unaffected by ACE inhibitors, potentially offering a mechanism for cardiac fibrosis progression despite ACE inhibition. Chymase might also participate in the fibrotic response by activating MMPs (Fang et al., 1997; Stewart et al., 2003). Both rodent and large animal studies on cardiac fibrosis emphasize the significance of mast cell chymases, suggesting potential therapeutic avenues. For instance, chymase inhibition reduced fibrosis and diastolic dysfunction in a dog model of tachycardia-induced heart failure (Matsumoto et al., 2003), decreased cardiac fibrosis and MMP expression in a porcine reperfusion infarction model (Oyamada et al., 2011), and attenuated interstitial fibrosis while preventing diastolic dysfunction in a rat non-reperfused myocardial infarction model (Kanemitsu et al., 2006). Tryptase, the most abundant product of human mast cells, effectively activates fibroblasts by promoting proliferation (Ruoss et al., 1991) and collagen I synthesis (Cairns and Walls, 1997) through the protease-activated receptor (PAR)-2, leading to ERK-MAPK signaling activation (McLarty et al., 2011). Despite in vitro evidence supporting the fibrogenic effects of tryptase and its expansion in fibrotic hearts (Frangogiannis et al., 1998b; Somasundaram et al., 2005), there is a lack of studies investigating the in vivo role of tryptases in fibrotic cardiac remodeling.
Although the available data generally indicate that mediators derived from mast cells play a role in the accumulation of fibrous tissue, certain experimental studies have proposed that mast cells might possess features that counteract fibrosis (Joseph et al., 2005). The specific processes responsible for this protective phenomenon are still not fully understood. A potential association may exist between the indirect effects of mast cells on the viability of cardiomyocytes or the expression pattern of growth factors (Ngkelo et al., 2016). Like macrophages, mast cells can modify the expression profile of growth factors and proteins in reaction to signals from the microenvironment and metabolic demand (Xiong et al., 2022). The data indicate a possible transition from a state that promotes fibrosis to one that inhibits fibrosis.
2.5 Dendritic cells in cardiac fibrosisDendritic cells (DCs) are novel players in various fibrotic diseases where they possess a central role as antigen-presenting cells to regulate the immune system and inflammatory response. Studies have reported an immunoprotective role of the infiltrated DCs in experimental post-MI healing (Nagai et al., 2014; Carvalheiro et al., 2020; Sun et al., 2021). A decreased number of DCs, associated with increased infiltration of macrophages, impaired reparative fibrosis, and cardiac rupture following MI, suggests a protective role of DCs in the cardiac healing process (Nagai et al., 2014). The myocardium harbors both conventional dendritic cells (cDC) and plasmacytoid dendritic cells (pDCs), which maintain the infiltration of other leukocytes in the injury site area. Infiltration of activated DCs into the injured myocardium mediates the regulation of monocyte and macrophage homeostasis in the infarct area (Naito et al., 2008). Depletion of bone marrow-derived CD11c+ cells resulted in sustained release of IL-1β, IL-18, TNF-α; a high level of MMP-9 activity; and a decreased level of IL-10 in a mouse model, which resulted in enhanced fibrosis (Anzai et al., 2012) (Figure 2).
Cardiac cDCs recruited by chemokine receptor CCR2 cause upregulation of cardiomyocyte hypertrophy and inflammation by advanced glycation end products (Cao et al., 2015). Santos et al. (2020) showed that tolerogenic DCs attenuated lymphoproliferation and increased the FOXP3+ CD4+ Treg cells in a mouse model. These events led to diminished IFN-γ, IL-12, and Col1a2 and increased IL-10, resulting in improved cardiac remodeling (Santos et al., 2020). These studies support the role of DCs in post-injury cardiac remodeling; however, more studies are needed to provide a clearer mechanistic insight into how DCs influence fibrosis.
3 Metabolic regulation of cardiac homeostasis3.1 Metabolic flexibility of the homeostatic heartThe normal adult heart derives approximately 70%–90% of ATP from the oxidation of fatty acids (FAs) and the remaining from the oxidation of glucose, lactate, ketone bodies, and certain amino acids. Mitochondrial oxidative phosphorylation generates most of the ATP required, whereas glycolysis and GTP formation in the TCA cycle provide only around 5% (Lopaschuk et al., 2010; Doenst et al., 2013). The coordinated FA oxidation enables the heart to maintain its ability to switch between available substrates. Moreover, FA produces the greatest ATP yield per 2-carbon energy substrates with the highest O2 consumption (Fillmore et al., 2014). On the other hand, glucose works as a flexible substrate as it provides ATP by cytoplasmic glycolysis and mitochondrial oxidation of the pyruvate derived from glucose metabolism. More importantly, glucose supplies ATP most efficiently in the ischemic and stressed myocardium (Tian et al., 2023). Glucose metabolism generates pyruvate, lactate, and acetyl-CoA, which are further used in the pentose phosphate pathway or replenish TCA cycle intermediates in the mitochondria. Thus, glucose plays a dynamic role in the overall cardiac metabolic balance.
Ketone bodies and amino acids have a minor contribution to the overall cardiac oxidative metabolism in a normal heart, but prolonged fasting, ketogenic diet, and poorly controlled diabetes increase the ketone body utilization by the heart in vivo, while lactate or ketone supplementation in the perfusate reduces the glucose and FA oxidation in isolated perfused hearts ex vivo (Jeffrey et al., 1995; Stanley et al., 2003; Wentz et al., 2010; Papazafiropoulou et al., 2021). Ketone bodies are readily metabolized by the heart, and β-hydroxybutyrate is predominantly oxidized in the heart. Ketones produce ATP with a median efficiency compared to FAs and glucose and hence are a major fuel for the heart with increased circulating ketone levels (Ho et al., 2021). On the other hand, the carbon skeleton produced on branched-chain amino acid (BCAA) metabolism, including acetyl-CoA, α-ketoglutarate, acetoacetyl CoA, succinyl CoA, pyruvate, fumarate, and oxaloacetate, is used in the processes of the TCA cycle to offset the energy demand (Newgard, 2012). The TCA cycle enzymes produce the reducing equivalents NADH and FADH2 within the mitochondrial matrix (Tymoczko et al., 2002) and act as a pivotal mechanism to interconnect the glycolytic, beta-oxidation, and amino acid oxidation pathways (Brandt U and Heinrich Pc, 2014).
Mitochondria not only generate energy but also contribute to cellular signaling, maintain redox equilibrium, and act as a hub for the interconnected metabolic pathways of different substrates (Schaper et al., 1985). Fatty acyl-coenzyme A (CoA) and pyruvate from FA and glucose metabolism, respectively, feed mitochondria, whereas lactate, ketone bodies, and amino acids get oxidized directly in the mitochondria. All the energy-yielding substrates converge on acetyl-CoA production via specific catabolic pathways, which ultimately enter the TCA cycle and accomplish the energy transfer through oxidative phosphorylation (Kolwicz et al., 2013; Passarella and Schurr, 2018). In short, the healthy adult heart is metabolically flexible, with FAs being the predominant substrate, followed by lactate, ketone bodies, glucose, and BCAAs (Ng et al., 2023). To meet its ATP demand for continual cardiac contraction and pumping activities, the heart can readily shift between different energy substrates.
3.2 Metabolic reprogramming in the stressed heartDuring the early events of fibrogenesis, mitochondrial and cellular homeostatic signaling and metabolic balance experience both qualitative and quantitative derangements. A shift in substrate preference away from FAs toward more anaerobic substrates leads the energy-compromised organ to suffer from a progressive burnout, which causes further functional deterioration (He et al., 2022). Perfused hypertrophic rat hearts showed a decrease in FA oxidation and increased glucose utilization consistently when subjected to regional myocardial infarction (Remondino et al., 2000), pressure overload (Allard et al., 1994; Christe and Rodgers, 1994), or volume overload (el Alaoui-Talibi et al., 1992; El Alaoui-Talibi et al., 1997) ex vivo. During stressful conditions, substrate versatility and complex regulatory mechanisms contribute to metabolic flexibility by transcriptional regulation and post-translational modification of crucial proteins in different metabolic pathways.
Limited oxygen supply during ischemia suppresses aerobic glucose and FA oxidation. The activation of the oxygen-sensing pathway and the HIF-1α leads to the transcriptional upregulation of glycolytic enzymes (Del Rey et al., 2017; Taylor and Scholz, 2022; Woods et al., 2022). Along with the shift of cardiac substrate metabolism from FA oxidation to glycolysis, GLUT4 gets translocated toward the sarcolemma, FA transporter FAT/CD36 moves away from the sarcolemma, and the glycogen content decreases (Heather et al., 2013). On the other hand, import of glucose into the cells by GLUT-4 depends on insulin, while glycogenolysis, triggered by an increase in AMP and inorganic phosphate and a decrease in ATP levels, generates glucose as an alternative source. Though glucose is not the major metabolic substrate for cardiac tissue, its utilization increases during ischemia, increased workload, and pressure overload hypertrophy (Stanley et al., 1992; Leong et al., 2002; Nascimben et al., 2004; He et al., 2012; Bertrand et al., 2020). This evidence supports metabolic reprogramming in cardiac cells during altered cardiac states as well as a shift in the metabolic axis, which is dependent not only on the energy demand but also on endocrine and neurohormonal homeostasis. Moreover, the heart relies more on glucose as its primary energy source in the context of disorders like diabetes and metabolic syndrome. This metabolic alteration has been found to have consequential effects on fibrosis-related cellular processes (Doenst et al., 2013; Hartupee and Mann, 2017).
Upregulation of ketone body utilization is another feature of the ischemic and hypertrophied failing heart to cope with the injurious event. 3-Hydroxybutyrate (3-OHB) enhances the bioenergetic thermodynamics of isolated mitochondria in the context of low FA levels. Moreover, a mouse model lacking 3-OHB oxidation is less adaptive to ischemic insult and pressure overload and culminated in worsened heart failure and remodeling (Aubert et al., 2016; Bedi et al., 2016; Horton et al., 2019; Manolis et al., 2023) However, more research is needed on cardiac ketone body metabolism in the context of pathological fibrosis and heart failure to elucidate the role of ketone bodies in the altered cardiac microenvironment.
Though amino acids have little contribution as oxidative fuel, myocardial uptake of several amino acids increases as a consequence of metabolic remodeling in pathological conditions. Amino acids are used in oxidative stress due to their potential non-oxidative metabolism and low contribution to cellular acidification. Glutamate and glutamine have been found to prolong cellular function when converted to α-ketoglutarate, while asparagine and aspartate remove amine groups and excess TCA cycle intermediates (Wischmeyer et al., 2003; Liu et al., 2007; Alkan and Bogner-Strauss, 2019). Moreover, glutamine and glutamate are used in ischemic and hypertrophied hearts to produce ATP directly through substrate-level phosphorylation and safeguard the cardiac tissue from being damaged by free radicals and low pH (Drake et al., 2012). Though the heart shows metabolic flexibility in terms of substrate preference governed by plasma substrate and hormone levels in normal physiological conditions, this phenomenon is significantly affected during pathology and failure (Jiang et al., 2021; Shi and Qiu, 2022). All these events are brought about as a result of the injury, metabolic adaptation to the altered microenvironment, and energy supply–demand mismatch and lead to changes in the ECM and architectural organization of the cardiac tissue and culminate in fibrosis.
3.3 How metabolism dyshomeostasis contributes to cardiac fibrosis?3.3.1 Direct effect of metabolism on the heart: cardiac fibroblasts and cardiomyocytesHighly regulated and interconnected networks of metabolic pathways not only provide the energy currency for the functional integrity of the heart but also maintain the structural and spatiotemporal homeostasis of the cardiac tissue. Different metabolic pathways perform predominant roles and orchestrate the background of metabolic reprogramming while adapting to different stages of physiological development or pathological conditions. Cardiac fibrosis is the endpoint of multifarious cardiovascular pathologies, such as ischemic and nonischemic heart failure, pressure and volume overloads, genetic cardiomyopathies, diabetes, and aging (Gibb et al., 2020).
The myocardium contains a complex and intricate consortium of cardiomyocytes, endothelium, fibroblasts, pericytes, and immune cells. Upon injury, these cells acquire a fibrogenic phenotype by upregulating the expression of fibrosis-related genes and exhibit matrix synthetic and remodeling profiles. Moreover, DAMPs from dead cardiomyocytes activate inflammation, and collectively with the inflammatory cytokines, TGF-β, and other mediators, these events contribute to cardiac fibrosis (Zeisberg et al., 2007; Zhang et al., 2015; Alex et al., 2023). However, correlative expression studies of ECM proteins attribute the cardiac fibroblasts as the primary cell type responsible for myocardial fibrogenesis. Cardiac fibroblasts comprise around 10%–20% of the total cell population in the heart (Pinto et al., 2016). The nature and activities of fibroblasts, their response to the altered microenvironment, and metabolic reprogramming define the course of cardiac remodeling following any insult (Ali et al., 2014; Kanisicak et al., 2016; Kaur et al., 2016).
Though quiescent cardiac fibroblasts derive energy from mitochondrial oxidative phosphorylation, activation of fibroblasts and their differentiation into myofibroblasts display a stark increase in aerobic glycolysis and lactate production (Gibb et al., 2020). Moreover, the early stages of cardiac fibroblast activation align with altered mitochondrial morphology (Xin et al., 2019), produce mitochondrial reactive oxygen species (mtROS) (Jain et al., 2013), and show features of mitochondrial Ca2+ uptake (Lombardi et al., 2019). Cardiac fibroblasts display remarkable plasticity in response to injurious stimuli, change their own behaviors, and adapt quickly to the altered environment by transitioning between differentiation states (Fu et al., 2020). Metabolic patterns are remodeled between the initiation of cardiac fibroblast activation and their full differentiation into myofibroblasts. Quiescent cardiac fibroblasts require ATP to acquire contractile phenotypes, while fully activated cardiac fibroblasts use amino acid synthesis for collagen production. Moreover, lactate, succinate, and other amino acids serve as stimulators of myofibroblast differentiation (Tian and Ren, 2023).
Altered glycolysis along with increased glycolytic enzymes, such as hexokinase, phosphofructokinase-1 (PFK1), pyruvate kinase, and lactate dehydrogenase (LDH), have been reported in activated cardiac fibroblasts and fibrotic diseases of different organs (Kottmann et al., 2012; Xie et al., 2015; Ding et al., 2017; Selvarajah et al., 2019). Following MI or angiotensin II administration, cardiac fibroblasts showed enhanced glycolysis and glutaminolysis, promoted the myofibroblast phenotype, and exacerbated myocardial injury (Lombardi et al., 2019). On the other hand, inhibition of glucose oxidation during enhanced glucose breakdown enables cardiac fibroblast activation and leads to the accumulation of lactate in myofibroblasts, which ultimately promotes histone lactylation following MI to express genes with a reparatory phenotype (Wang et al., 2022). Furthermore, increased levels of lactate enhance cardiac fibrosis and worsen cardiac dysfunction by promoting endothelial-to-mesenchymal transition via the transcription factor Snail1 lactylation after MI (Fan et al., 2023). Myofibroblasts undergo a shift away from FA oxidation toward glutamine utilization and α-ketoglutarate (α-KG) production. Enhanced glutaminolysis augments α-KG biosynthesis and cardiac fibroblast activation and contributes to de novo collagen synthesis from differentiated myofibroblasts (Gibb et al., 2022a).
3.3.2 Direct effect of metabolism on nonmyocytes: immune cellsAlthough the cardiomyocyte is the heart’s parenchymal cell, the
留言 (0)