While soft-tissue xanthomas are relatively common lesions associated with accumulation of cholesterol-rich material in hyperlipidemic patients, its bony counterpart is rare [1, 2]. A recent advancement in our understanding of lipid protein metabolism has enabled us to determine that circulating lipoproteins increasingly deposit into adjacent tissues following local trauma or hemorrhage. The cholesterol passes between the vascular endothelial cells into different sites of the body as non-degradable sterols that are later eliminated by tissue macrophages through phagocytosis [3]. This process of elimination gives rise to “foamy cells” or xanthoma cells, a characteristic feature of xanthomas. Additionally, it has been suggested that these lesions could be manifestations of a pre-existing pathological condition, such as degenerative alterations associated with different types of lesions, including aneurysmal bone cyst, traumatic bone cavity, fibrous dysplasia, or giant cell tumors [4, 5].
The central or intraosseous xanthomas that particularly affect the appendicular and axial bones are typically linked to lipid disorders associated with endocrine or metabolic diseases [7]. A review of the current literature suggested very few patients had systemic diseases associated with intraosseous xanthomas. Brooks et al. 2017 described xanthoma of the jaw bones (XJB) in a 16-year-old male with hyperlipidemia and vitamin D deficiency [8]. Another case was reported of a 15-year-old male having XJB in conjunction with Noonan Syndrome, an autosomal dominant disorder that is associated with the development of giant cell lesions in the jaws [9]. Other studies outlined the connection of XJB with conditions such as diabetes and orofacial granulomatosis [6, 10]. The etiology of non-systemic XJB is currently unknown. A recent study reported the lack of a targeted DNA mutation in intraosseous xanthomas after investigating 50 of the most common cancer-related genes [9]. Due to lack of certainty in etiopathogenesis, intraosseous xanthomas are thought to be caused by a reparative or reactive process of a previously diagnosed pathological condition [10,11,12].
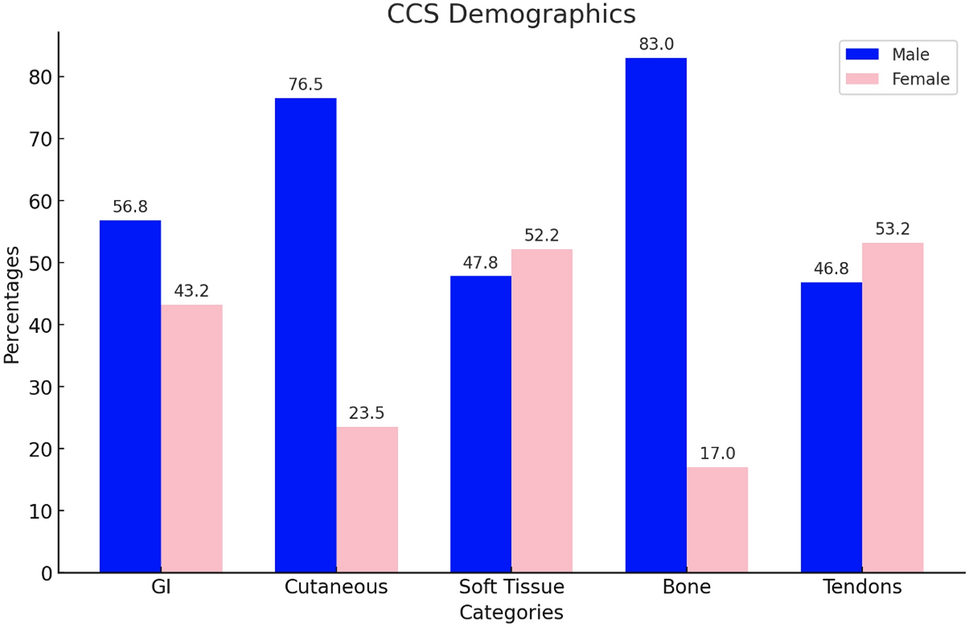
Previous reports have suggested that XJBs occur more commonly in males [11]. However, our comprehensive analysis showed a slight female predilection (44.4%, n = 20 males and 55.6%, n = 25 females), in contrast to the previous reports and the findings of this study. The age range of patients affected with XJB is from 11 to 72 years of age, with an average age of 30.01 years which aligns with the present study [12]. No age differences between women and men were noted. Although 90% of the cases affected the mandible, recent publications described 4 cases occurring in the maxilla. XJB in the maxilla represents only 7.4% of all the cases reported [11, 13, 14]. In 32.1% (n = 17) of cases, XJBs demonstrated swelling, tooth mobility or displacement [2, 4, 6, 8,9,10,11, 13,14,15,16]. Importantly, a large proportion of cases exhibited cortical erosion or perforation, and resorption of roots. In some cases, the increased bony expansion caused considerable destruction of the jaw. The associated or adjacent teeth are almost always vital with little to no tenderness on percussion. Pain was an uncommon finding, and almost 90% of the cases were detected during routine radiographic examination as in our cases. Dull pain and mucosal numbness were the most common findings in symptomatic cases [9, 11, 13, 15].
XJBs exhibit radiographic features that overlap with many common lesions of the jaw, however, cone-beam computed tomography (CBCT) imaging may offer a helpful diagnostic tool [14]. Based on the available literature, the majority of XJBs show multilocular or unilocular, punched out, radiolucent lesions with or without sclerotic borders. Although several studies, including the present study, have reported mixed and even radiopaque radiographic presentation [2, 3, 8, 10, 14, 15]. Harsanyi et al. reported seven cases of XJB, two of which exhibited a ground glass appearance [2]. This variation may be attributed to reactive bone formation (Fig. 2-c), dystrophic calcification, or the deposition of calcified material, similar to that seen in fibrous dysplasia [16].
The radiographic characteristics are not definitive for diagnosing XJB but suggest an initial differential diagnosis of lesions such as odontogenic tumors/cysts or periapical inflammation [14]. Therefore, histopathological and immunohistochemical analyses are essential for diagnosis. The dominant feature of this entity is the presence of sheets of lipid-laden histiocytes (xanthoma cells), which are also known as foamy cells, with abundant cytoplasm and small hyperchromatic nuclei. These cells are surrounded by fibrous connective tissue with no significant inflammation. Other histological findings include the presence of giant cells, lamellar and woven bone fragments and dystrophic calcifications [11, 12].
Diagnosing XJB histopathologically can be challenging, especially in secondary inflamed cases. Other intraosseous lesions, such as non-ossifying fibroma (NOF) and benign fibrous histiocytoma (BFH), also display focal collections of foamy histiocytes and multinucleated giant cells. These cells are distributed within a fibrous connective tissue stroma that often demonstrates a whorling or storiform pattern [11, 13]. Granular cell odontogenic tumor (GCOT) also falls in the list of xanthomatous lesions. Histologically, GCOTs demonstrate islands or strands of inactive odontogenic epithelium interspersed within a background of granular mesenchymal cells. The absence of both fascicular fibrous stroma and odontogenic epithelium islands aids in distinguishing XJB from other intraosseous xanthomatous lesions. Reactive foamy histocytes are also seen in periapical inflammatory lesions. Yet, these histocytes are often intermixed with varying numbers of chronic and/or acute inflammatory cells and should not be interpreted as XJB [3]. Nonetheless, clinical presentation (i.e., non-vital teeth) can aid in establishing a definitive diagnosis. Other disorders that present with xanthomatous cells include Rosai-Dorfman, Erdheim-Chester, and Gaucher diseases. Gaucher disease, the most common lysosomal storage disease, can also affect the gnathic bones [17]. In Gaucher disease, a genetic mutation leads to the accumulation of lipids in various organs, including bones. The histopathologic exam demonstrates vacuolated, lipid-laden reticuloendothelial cells (Gaucher’s cells) infiltrating the connective tissue, characterized by enlarged, granular cytoplasm and round, displaced nuclei [17]. Furthermore, systemic metabolic and lipid diseases, such as type II and III hyperlipidemia and diabetes mellitus, should be included for a complete differential diagnosis [15]. Hyperlipidemia, a condition characterized by elevated levels of lipids (fats) in the blood, is categorized into different types based on specific lipid profiles [18]. Elevated lipid levels can lead to the accumulation of fats in tissues and may cause subcutaneous and tendon xanthomas, xanthelasma (cholesterol deposits building up under the skin), and corneal arcus (deposition of lipid in the peripheral cornea). However, bony manifestations are uncommon, but can be the first sign of the disease [15, 19]. Unlike intraosseous xanthomas, the foamy macrophages associated with hyperlipidemia are associated with cholesterol crystal clefts, inflammatory reactions, and giant cells, which can result in fibrosis [15]. Since these diseases frequently show histopathological overlap, an immunohistochemical analysis as well as evaluation of pertinent medical history is important to distinguish them from XJB [15].
Immunohistochemically, xanthomas demonstrate strong cytoplasmic positivity for CD68, a marker used to identify the activated macrophages [3]. Besides CD68, supplementary markers such as S100, factor XIIIa + and CD1a are useful in separating xanthomas from other diseases. These three markers help identify neural cells, histiocytes and Langerhans cells respectively [15]. It is observed that both S100 and CD68 are diffusely positive in Rosai-Dorfman disease, Erdheim disease and Gaucher disease. Similarly, S100 and CD1a are consistently positive for Langerhans cell histiocytosis thus making them reliable in distinguishing xanthomas from Langerhans disease. Although both NOF and BFH exhibit variable positivity to CD68, they are strongly positive for factor XIIIa + making this immunohistochemical tool important in differentiating tumors with fibrohistiocytic lineage from xanthoma of bone [3, 11, 15].
XJB generally has an excellent prognosis. Though XJB can cause extensive bone destruction, curettage is the widely accepted method of treatment. This study is consistent with most reported cases, which demonstrated complete healing after surgical intervention. However, of the 46 cases reported in the literature, only 5 cases exhibited progression [2, 10]. Research indicates that curettage of XJB lesions is effective in promoting bone regeneration [3]. Interestingly, Saha et al. presented a case of XJB where initial surgical removal of the lesion was followed by a second surgical intervention to completely remove the lesion [20]. During the second procedure, newly formed woven bone with absence of xanthoma cells was noted in the central portion of the lesion, supporting adequacy of a single surgical intervention.






留言 (0)