
記住我


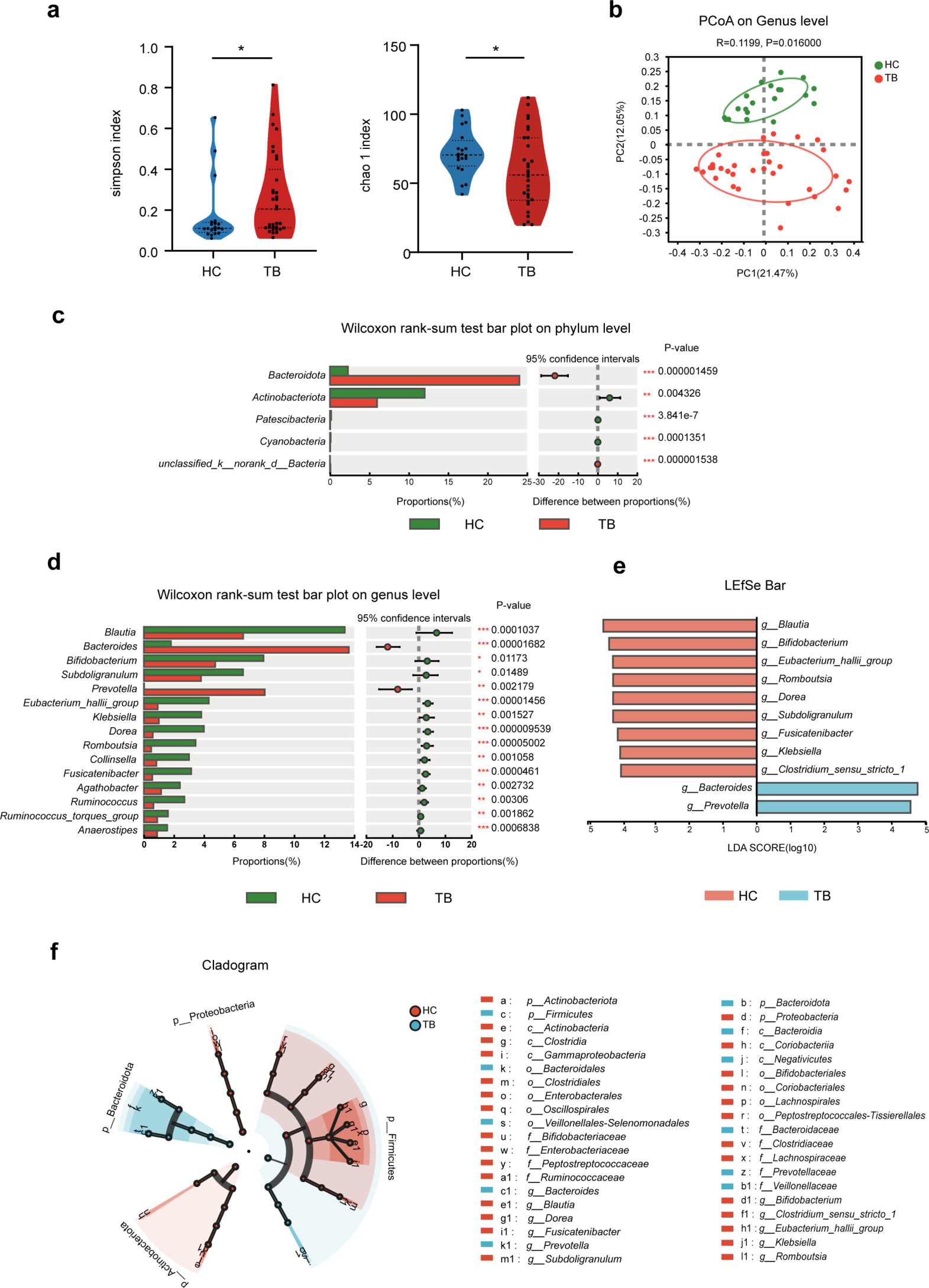


In this study, we collected three tissue samples each from the control and diabetic groups, referred to as WT-TC and DB-TC, respectively. After generating the gene expression matrix, we identified 449 upregulated and 1,006 downregulated differentially expressed genes (DEGs) (Fig. 1A). Functional enrichment analysis of the DEGs showed that they were mainly associated with metabolism-related Gene Ontology (GO) terms, such as lipid and monocarboxylic acid metabolic processes (Fig. 1B). KEGG pathway analysis revealed that the DEGs were enriched in metabolic pathways such as the PPAR signaling pathway and retinol metabolism (Fig. 1C). Moreover, Reactome pathway analysis demonstrated that the DEGs were also enriched in various metabolic pathways, including Metabolism of lipids and Fatty acid metabolism (Fig. 1D). To obtain protein-protein interaction (PPI) networks, the DEGs were analyzed using the Metascape website, and the resulting networks were visualized (Fig. 1E). The MCODE plug-in in Cytoscape was used to identify characteristics of each node in the network graph, and the largest subnetworks were selected and visualized (Fig. 1F). MCODE1 functions were mainly enriched in the metabolism of lipids and response to vitamins.
Fig. 1
Differential expression and functional analysis of genes in control and diabetic groups. (A) Volcano plot showing the differentially expressed genes in control (WT-TC) and diabetic (DB-TC) groups. 449 genes were upregulated (red) and 1,006 genes were downregulated (blue). The x axis is log2 (Fold Change), which is the multiple of the difference between two groups of samples and the y axis showed the log P-value which calculated by t-test. The cut-off is 1.3=-log10 (0.05). (B) Gene Ontology (GO) functional enrichment analysis of differentially expressed genes. The top enriched GO terms were related to metabolism, such as the lipid metabolic process and the monocarboxylic acid metabolic process. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes. The top enriched pathways were metabolic pathways, such as the PPAR signaling pathway and retinol metabolism. (D) Reactome pathway analysis of differentially expressed genes. The top enriched pathways were related to various metabolic processes, such as the metabolism of lipids and fatty acid metabolism. (E) Protein-protein interaction (PPI) network analysis of differentially expressed genes using Metascape. The network was visualized with gene information, and nodes were colored based on their degree of connectivity. (F) The largest subnetworks in the PPI network were selected and visualized using the MCODE plug-in in Cytoscape. MCODE1 functions were mainly enriched in the metabolism of lipids and the response to vitamins
16 S rDNA sequencing results and changes in microbial diversityTo verify the reasonableness of the sample size, we examined the dilution curve and rank abundance curve. The dilution curve (Fig. 2A) showed that the results tended to become more reasonable as the sample size increased. The rank abundance curve (Fig. 2B) indicated that the conjunctival flora’s abundance was higher and more evenly distributed in the WT-TC and DB-TC groups, but the DB-TC group’s abundance was lower than that of the WT-TC group.
Fig. 2
Analysis of microbial diversity in control and diabetic groups using 16 S rDNA sequencing. (A) Dilution curve showing the reasonableness of the sample size with the increase of sample size. The x axis represents the number of sequencing samples randomly selected from a certain sample, and the y axis represents the number of OTUs that can be constructed based on this number of sequencing samples to reflect the depth of sequencing. (B) Rank abundance curve showing the abundance and evenness of conjunctival flora in the WT-TC and DB-TC groups. (C) Alpha diversity indices including Pielou, Shannon, and Simpson indices, reflecting the abundance and diversity of microbial communities in the samples. The overall index of the WT-TC group was higher than that of the DB-TC group, indicating a higher degree of microbial community diversity in the control group. The occurrence of diabetes caused a decrease in species richness and diversity (p < 0.05). The goods_coverage index for each group was greater than 0.99, indicating high credibility of the data
Alpha diversity reflects the diversity of species in a single sample, and various indices such as Pielou index, Sobs index, Chao index, and ACE index reflect the community’s abundance, while the Shannon index, PD index, and Simpson index reflect the community’s diversity. The goods_coverage index reflects the sequencing depth, and a higher value indicates higher credibility. The goods_coverage index for each group was greater than 0.99, indicating highly credible data. Alpha diversity indices (Fig. 2C) with significant differences under different conditions were screened by a rank sum test, including the Pielou, Shannon, and Simpson indices. The overall index of the WT-TC group was higher than that of the DB-TC group, indicating higher microbial community diversity in the former. Diabetes caused a decrease in species richness and diversity (p < 0.05).
mBeta diversity was used to compare the magnitude of differences between pairs of samples in terms of species diversity. Bray-Curtis, weighted UniFrac, and unweighted UniFrac were used. Figure 3A is the beta diversity matrix heat map. The beta diversity data were visualized graphically, and the samples were clustered by clustering samples with similar beta diversity together, reflecting the similarity between samples. As seen from the figure, the respective beta diversity between the two groups was well clustered, and the groups could be clustered within a certain range. Based on the beta diversity distance matrix information, the samples were classified into UPGMA classification trees. The cluster analysis results for all samples are shown in Fig. 3B, indicating that the WT-TC and DB-TC groups have similar colony results but differ in compositional abundance.
Fig. 3
mBeta diversity was used to compare the magnitude of differences between pairs of samples in terms of species diversity. (A) The beta diversity matrix heat map obtained using Bray-Curtis, weighted UniFrac, and unweighted UniFrac analyses. Samples with similar beta diversity were clustered together, revealing the similarity between them. The beta diversity between the two groups was well-clustered and could be distinguished within a certain range. Based on the beta diversity distance matrix, the samples were classified into UPGMA classification trees, where the more similar samples had shorter common branches. (B) The results of cluster analysis for all samples, indicating that the WT-TC and DB-TC groups had similar colony results but differed in their compositional abundance
Elaborating the distribution of bacterial flora through 16 S rDNA sequencingAt the phylum level, the top 10 phyla of conjunctival flora in both groups of mice were Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, Crenarchaeota, Chloroflexi, Cyanobacteria, Patescibacteria, Planctomycetes, and Nitrospirae. The four predominant phyla were Proteobacteria, Firmicutes, Bacteroidetes, and Cyanobacteria (Fig. 4A). Compared to the control group, the abundance of Firmicutes increased in the diabetic group, while the proportions of Proteobacteria, Bacteroidetes, and Cyanobacteria decreased significantly (p < 0.05) (Fig. 4B).
Fig. 4
Microbial diversity in conjunctival flora of diabetic and control mice. (A) Relative abundance of the top 10 phyla, classes, orders, families, genera, and species in both groups of mice. (B) Comparison of the abundance of major phyla, classes, orders, families, genera, and species between diabetic and control mice
At the class level, the top 10 classes of conjunctival flora in both groups of mice were Gammaproteobacteria, Bacilli, Bacteroidia, Alphaproteobacteria, Actinobacteria, Bathyarchaeia, Oxyphotobacteria, Patescibacteria, Clostridia, and Deltaproteobacteria. The five dominant classes were Gammaproteobacteria, Bacilli, Bacteroidia, Alphaproteobacteria, and Actinobacteria (Fig. 4A). Compared to the control group, the abundance of Bacilli increased in the diabetic group, while the abundance of Gammaproteobacteria, Bacteroidia, Alphaproteobacteria, and Actinobacteria proportions were significantly reduced (p < 0.05) (Fig. 4B).
At the order level, the top 10 orders of conjunctival flora in both groups of mice were Enterobacteriales, Lactobacillales, Chitinophagales, Bacillales, Betaproteobacteriales, Corynebacteriales, Rickettsiales, Rhizobiales, Vibrionales, and Pseudomonadales (Fig. 4A). Compared to the control group, the abundance of Lactobacillales, Pseudomonadales, and Bacillales increased in the diabetic group, while the proportion of Rickettsiales, Rhizobiales, Enterobacteriales, Chitinophagales, and Betaproteobacteriales were significantly reduced (p < 0.05) (Fig. 4B).
At the Family level, the top 10 families in terms of relative abundance of conjunctival flora in both groups of mice were Enterobacteriaceae, Enterococcaceae, Chitinophagaceae, Staphylococcaceae, Burkholderiaceae, Corynebacteriaceae, Rickettsiaceae, Vibrionaceae, Xanthobacteraceae, and Moraxellaceae (Fig. 4A). Compared to the control group, the abundance of Staphylococcaceae, Moraxellaceae, and Enterococcaceae increased in the diabetic group, while the proportions of Xanthobacteraceae, Rickettsiaceae, Enterobacteriaceae, Chitinophagaceae, and Burkholderiaceae were significantly reduced (p < 0.05) (Fig. 4B).
At the Genus level, the top 10 genera in terms of relative abundance of conjunctival flora in both groups of mice were Enterococcus, Sediminibacterium, Staphylococcus, Ralstonia, Corynebacterium_1, Rickettsia, Rosenbergiella, Pantoea, Vibrio, and Bradyrhizobium (Fig. 4A). Compared to the control group, there was an increase in the abundance of Staphylococcus, Rosenbergiella, Moraxellaceae, and Enterococcus in the diabetic group, while the proportions of Sediminibacterium, Rickettsia, Ralstonia, Pantoea, and Bradyrhizobium were significantly reduced (p < 0.05) (Fig. 4B).
At the Species level, the top 10 species in terms of relative abundance of conjunctival flora in both groups of mice were Enterococcus_faecalis, Staphylococcus_sciuri, Corynebacterium_glutamicum, Corynebacterium_sp_CNJ-954, Pantoea_dispersa, Proteus_vulgaris, Acinetobacter_lwoffii, Rubrobacter_sp_LYG58, Tsukamurella_pulmonis, and Chryseobacterium_indologenes (Fig. 4A). Compared to the control group, the abundance of Staphylococcus_sciuri, Proteus_vulgaris, Enterococcus_faecalis, and Acinetobacter_lwoffii increased in the diabetic group, while the proportions of Tsukamurella_pulmonis, Pantoea_dispersa, and Corynebacterium_sp_CNJ-954 were significantly reduced (p < 0.05).
ITS sequencing results and changes in microbial diversityThe dilution curve (Fig. 5A) shows that the results become more reliable with increasing sample size. The abundance rank curve (Fig. 5B) suggests that conjunctival flora in both the WT-TC and DB-TC groups are more abundant and evenly distributed than in the control group. The abundance of conjunctival flora is lower in the DB-TC group than in the WT-TC group. Alpha diversity analysis did not show any statistically significant difference in fungal species abundance and diversity between the two groups (p > 0.05), indicating that diabetes did not significantly impact the abundance and diversity of fungal species (Fig. 5C). Beta diversity analysis showed poor beta diversity clustering between the two groups (Fig. 6A), and the clustering analysis of all samples indicated that the fungal colony composition of the WT-TC and DB-TC groups did not significantly differ (Fig. 6B).
Fig. 5
ITS sequencing results. (A) The dilution curve of conjunctival flora samples, which indicates a reasonable increase in results with increasing sample size. The x axis represents the number of sequencing samples randomly selected from a certain sample, and the y axis represents the number of OTUs that can be constructed based on this number of sequencing samples to reflect the depth of sequencing. (B) The abundance rank curve of the WT-TC and DB-TC groups, revealing that the abundance of conjunctival flora in the two groups is higher and more evenly distributed, while the abundance of the DB-TC group is lower than that of the WT-TC group. (C) Alpha diversity analysis revealed no statistical difference in fungal species abundance and diversity between the two groups (p > 0.05)
Fig. 6
Results of Beta diversity analysis. (A) Using clustering analysis, poor clustering was shown between the two groups. (B) The fungal colony composition of the WT-TC and DB-TC groups. There are no significant differences between all groups, as indicated by the clustering analysis. These findings suggest that the occurrence of diabetes did not impact the fungal colony composition in the conjunctiva
Elaborating the distribution of fungal mycota by ITS sequencingTable 1 presents the results of ITS sequencing for the two groups of samples. Most of the fungi identified in the samples belonged to the phylum Ascomycota, class Sordariomycetes, order Sordariales, family Bolbitiaceae, and genus Agrocybe. At the species level, the highest frequency OTU1 matched four different fungal ITS regions: Agrocybe pediades, Serendipita indica, Humicola grisea, and Rhizopus arrhizus.
Table 1 The results of ITS sequencing for the two groups of samples. Most of the fungi identified in the samples belonged to the phylum Ascomycota, class Sordariomycetes, order Sordariales, family Bolbitiaceae, and genus Agrocybe. At the species level, the highest frequency OTU1 matched four different fungal ITS regions: Agrocybe pediades, Serendipita indica, Humicola grisea, and Rhizopus arrhizusFurthermore, the abundance of Eurotiomycetes, Eurotiales, Mucoraceae, and Mucor was significantly lower in the diabetic group compared to the control group (Fig. 7, p < 0.05).
Fig. 7
The fungal taxonomic composition and abundance differences between the WT-TC and DB-TC groups based on ITS sequencing. The taxonomic composition of the fungal community at the phylum, class, order, family, and genus levels is presented. Ascomycota, Sordariomycetes, Sordariales, Bolbitiaceae, and Agrocybe were the dominant taxa found in the samples. At the species level, the most high-frequency OTU1 was identified as Agrocybe pediades, Serendipita indica, Humicola grisea, and Rhizopus arrhizus. The abundance of Eurotiomycetes, Eurotiales, Mucoraceae, and Mucor was significantly reduced in the diabetic group compared to the control group (p < 0.05)
Functional prediction of 16 S rDNA and ITS sequencing communitiesTax4Fun software was used to perform functional annotation of KEGG pathways based on the species annotation and abundance information of OTUs. The abundance information of each pathway and KO ID was counted, and 16 KEGG secondary functional pathways with relatively high abundance are shown in Fig. 8A. The predicted functional analysis revealed that all samples were related to carbohydrate, capacity, lipid, terpene and polyketides, amino acid metabolic pathways, biodegradation and metabolism, environmental adaptation, digestion, circulatory and excretory systems, cancer and immune diseases, and cellular process pathways of cell growth and death.
Fig. 8
Functional prediction of 16 S rDNA and ITS sequencing communities. (A) The predicted functional analysis of the KEGG pathways for all samples using Tax4Fun software. The top 16 KEGG secondary functional pathways with relatively high abundance are displayed, including pathways related to carbohydrate, capacity, lipid, terpene and polyketides, amino acid metabolic pathways, biodegradation and metabolism, environmental adaptation, digestion, circulatory and excretory systems, cancer and immune diseases, and cellular process pathways of cell growth and death. (B) The relative abundance of 22 fungal functional taxa (excluding unassigned taxa) analyzed using the FUNGuild tool. The functional taxa are ranked by abundance, and the results indicate that “Bryophyte Parasite-Dung Saprotroph-Ectomycorrhizal-Fungal Parasite-Leaf Saprotroph-Plant Parasite-Undefined Saprotroph-Wood Saprotroph” had the highest abundance among all samples, followed by undefined saprotroph and then orchid mycorrhizal
Additionally, a predicted functional analysis of all treated fungal communities was conducted using the FUNGuild tool. Figure 8B shows the relative abundance of 22 fungal functional taxa (excluding unassigned taxa), and the results indicated that “Bryophyte Parasite-Dung Saprotroph-Ectomycorrhizal-Fungal Parasite-Leaf Saprotroph-Plant Parasite-Undefined Saprotroph-Wood Saprotroph” had the highest abundance among all samples, followed by undefined saprotroph and then orchid mycorrhizal.
Comparison of bacterial and fungal diversityTo assess the consistency of bacterial and fungal richness levels, correlation coefficients were used to analyze the trends of bacterial and fungal species in each subgroup at the species diversity (alpha diversity) level. Correlation scatter plots were then created to visualize the effect of correlation. The results indicated that there was no significant correlation between changes in bacterial and fungal species diversity, suggesting that bacteria and fungi responded differently to grouping differences (Fig. 9A). At the beta diversity correlation level, statistical tests were performed for within-group and between-group distance differences, and the combined distance box plot showed that grouping differences affected bacteria at the Phylum, Order, Family, Genus, and Species levels, while fungi were only affected at the Order, Family, and Genus levels (Fig. 9B).
Fig. 9
Comparison of bacterial and fungal diversity. (A) The correlation scatter plots of bacterial and fungal species diversity indices, indicating no significant correlation between the two at the alpha diversity level. (B) The statistical test results for within-group and between-group distance differences, as well as the combined distance box plot, demonstrating that grouping differences affected bacteria at the Phylum, Order, Family, Genus, and Species levels and fungi only at the Order, Family, and Genus levels. (C-E) The correlation analyses between bacteria and fungi at the family, genus, and OTU levels using the Sparse Correlations for Compositional data model. The heat map and circos plots are based on the sPLS screening of strongly correlated species, with the internal circos presenting correlation results and the external heat map displaying specific species abundance trends. The results show that there were correlations between bacteria and fungi at all three taxonomic levels, indicating interactions between bacteria and fungi
The Sparse Correlations for Compositional data model was used to analyze the correlations between bacteria and fungi at different taxonomic levels (Fig. 9C and E). Species with mean values of abundance greater than 0.1% at the family, genus, and OTU levels were screened for correlation analysis. A combined heat map and circos were plotted based on the sPLS screening of strongly correlated species. The internal circos presented the correlation results, including positive and negative correlations and the intensity of the correlation. The external heat map showed the specific species abundance trends. The results showed that there were correlations between bacteria and fungi at the family, genus, and OTU levels, indicating that there were some interactions between bacteria and fungi.
留言 (0)