
記住我


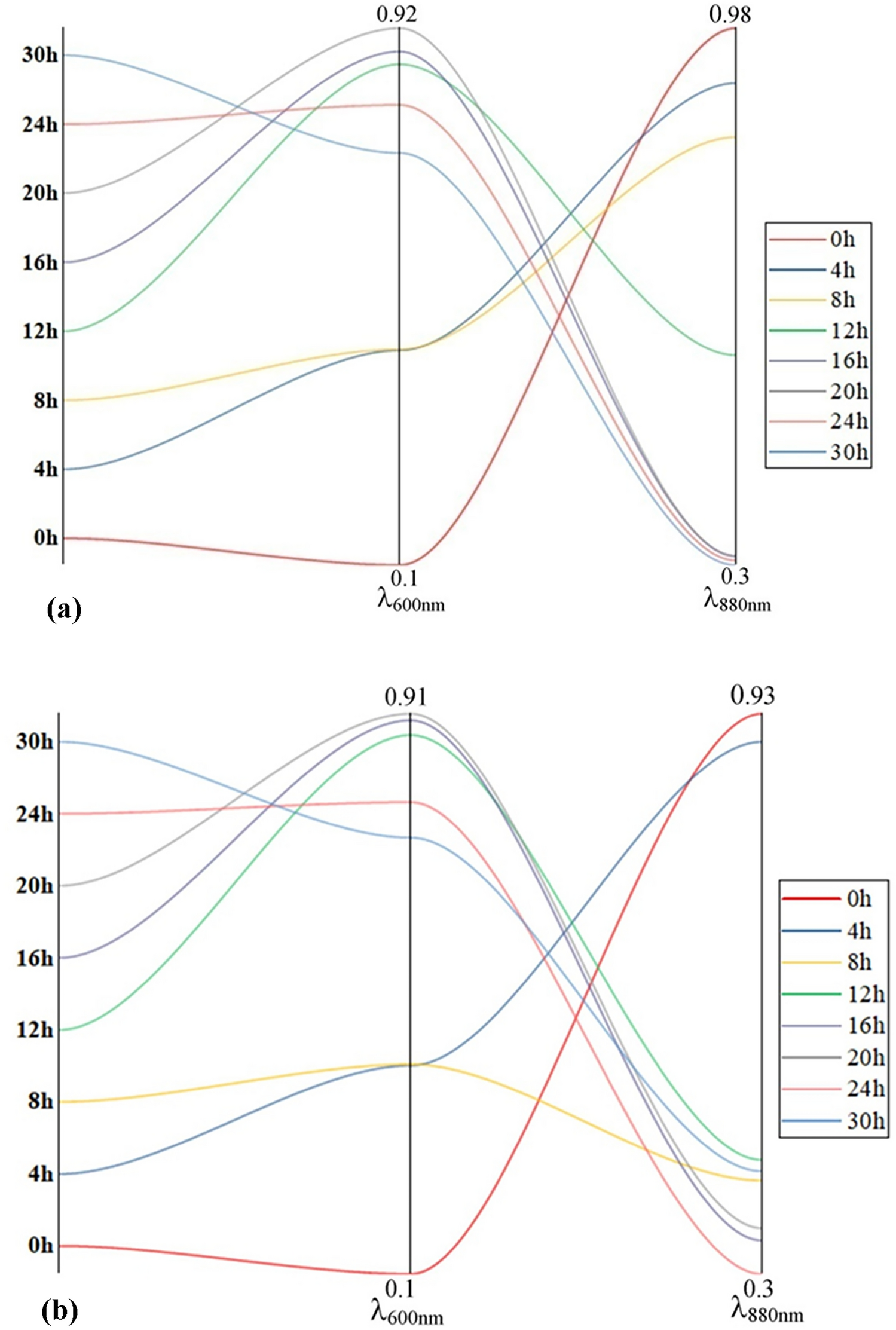





The sequences of fungal/algal 18S rRNA gene (near-full-length) from the sampled lichens in Austria and Uganda were BLAST-searched. The analysis of fungal partners across all 16 samples indicated that they all belonged to the genus Umbilicaria (fall under the Ascomycota phylum), and matching rates of 99.30% or greater were observed, along with query coverages of 97% or higher (Table S2). The sequence similarity between the Austrian and Ugandan mycobionts ranged from 99.18% to 99.88%. The sequences of fungal 18S rRNA gene (near-full-length), only ITS regions, and ITS regions with elongation from the sampled lichens in Austria and Uganda were BLAST-searched (Table S2–S4). The analysis of fungal partners across all 16 samples indicated the closest relationship to the Umbilicaria rhizinata voucher agrED295 or U. aprina voucher agrED360, with matching rates of 99.30% or greater (Table S2), the sequence similarity between the Austrian and Ugandan 18S rRNA gene ranged from 99.18% to 99.88%; U. africana voucher acpED473, U. aprina isolate AFTOL-ID 7153 and U. aprina isolate AFTOL-ID 7116 with all the same matching rates of 99.79% (Table S3); U. africana voucher acpED473, U. aprina isolate AFTOL-ID 7153 and U. aprina isolate AFTOL-ID 7116 with matching rates of 98.70% or greater (Table S4); determined by the BLAST-searched results of sequences of fungal 18S rRNA gene (near-full-length), only ITS regions, and ITS regions with elongation, respectively. It should be noted that although in the BLAST results, the highest similarities of all sequences are divided into three different species: U. rhizinata, U. aprina, and U. africana. However, U. rhizinata is a synonym of U. aprina, and U. africana was also classified in the U. aprina group [2, 7]. Therefore, according to the current classification, all of them can be collectively classified as the U. aprina group. The more detailed classification requires further research.
The examination of algal partners within the 16 samples revealed their closest affiliation to the green algal lineage Trebouxia jamesii (UBT-86.132E2), a widely observed photobiont in lichens [45], and matching rates of 98.46% or greater were observed in the Uganda region (Table S7). But in the Austrian samples only similarity values between 96.85% and 97.67%. The sequence similarity between the Austrian and Ugandan photobionts ranged from 96.09% to 98.26%.
Evaluation of MiSeq-generated V3-V4 Sequences and OTUsBy utilizing Illumina MiSeq sequencing, a cumulative sum of 755,198 raw reads was initially generated from a total of 16 collected target lichen specimens. After undergoing filtering procedures, 572,363 valid paired reads remained, and these were subsequently organized into OTUs. Drawing from the analysis data present in the EzBioCloud database [33], the average length of all valid reads stood at 403.0 base pairs (bp). The sequences from the Austrian samples exhibited an average length of 404.2 bp, while those from the Ugandan samples displayed a slightly shorter mean length of 402.4 bp. Table 3 provides comprehensive details encompassing raw reads, valid reads, the counts of grouped OTUs, species, genera, families, orders, classes, and phyla derived from OTUs, as well as the mean sequence length for each sample.
Table 3 Each composition of lichen samples is characterized by the counts of MiSeq-generated V3-V4 region reads, 97% similarity-based OTUs, OTU-derived species, genera, families, orders, classes, and phylaThe coverage observed in the rarefaction analysis signifies the ratio between the actual number of observed OTUs and the estimated OTU count. This equivalence aligns with the alpha-diversity index, Chao1. The coverages in Alpine lichen samples can be calculated from Table 5 and mean/minimum/maximum ratios were 91.75%/85.26% (in U9)/97.48% (in A02), respectively. The coverage ratios suggested valid reads generated in this study are deemed adequate for subsequent statistical and bioinformatic analyses.
Table 4 displays the distribution of taxa counts across different regions (including OTUs, species, genera, families, orders, classes, and phyla) that were exclusively identified in Austrian lichen samples, those found solely in Ugandan samples, and those observed in samples from both regions. Observed OTUs, species, and genus ranks showed higher percentages of region-specific features, while the order, class, and phylum ranks were shown in both regions with more than half of region-common features. The findings revealed a similarity between the two regions at higher taxonomic ranks, accompanied by distinctiveness unique to each region at lower taxonomic levels.
Table 4 The counts of assigned OTUs, as well as OTU-derived species, genera, families, orders, classes, and phyla, are detailed for the categories of solely Austrian, and solely Ugandan, and shared across both regionsTaxonomic Composition of Lichen-associated Bacterial CommunitiesFigure 2 presents the compositions of bacterial phyla, analyzed from the OTUs within the collection of 16 lichen samples. In total eight phyla of bacteria were found to be common features with read frequencies of > 1% in all 16 samples. Each lichen sample contained 8–20 bacterial phyla (Table 3) including 5–15 phyla less than 1% of read frequencies. According to modern nomenclature, The popular phyla were sorted by name alphabetically as follows: Acidobacteriota, Actinomycota, Armatimonadota, Bacteroidota, Chloroflexota, Deinococcota, Planctomycetota, and Pseudomonadota.
Fig. 2
The bacterial compositions on phylum rank of OTUs, derived from collected target lichen samples collected in Austria (A01 to A05) and Uganda (U1 to U11), are depicted. A total of eight different phyla were identified with read abundances exceeding 1% of the total read count for each region. Compositions of bacterial classes, orders, families, genera, and species are shown in Figures S3–S6
Alpha and Beta DiversityTo assess the OTU richness of each collected target lichen sample, alpha-diversity indices were employed, encompassing computations for Chao1 richness, Shannon indices, and Simpson indices. These computed results are presented in Table 5. Chao1 richness values and Shannon/Simpson indices were used for estimating OTU numbers for the rarefaction curve analyses and calculations of the effective number of species (ENS), respectively [46]. Chao1, Shannon, ENS values and observed OTU numbers have positive correlations with species and evenness, and Simpson index values have negative correlations with species and evenness. As a result, the Austrian samples exhibited elevated Chao1, Shannon, and ENS values, alongside a greater count of observed OTUs, and lower Simpson index values. These patterns collectively indicate higher species richness and evenness within the Austrian samples. However, it is noteworthy that the Ugandan sample U10 displayed the highest values across these metrics, as illustrated in Table 3.
Table 5 Regarding the OTUs of bacteria obtained from five collected target lichen samples sourced in Austria (designated A01 to A05) and 11 samples from Uganda (labeled U1 to U11), alpha-diversity indices encompassing Chao1 richness, Shannon indices, and Simpson indices were computedDue to different calculation methods, Shannon/Simpson indices cannot be used to estimate bacterial species richness. Comparatively, Chao1 values were close to estimated OTU numbers and may better represent species richness in the case of large sample sizes, as reported in other studies [47, 48].
Beta diversity was used for evaluating similarity/dissimilarity between different samples including PCA and hierarchical cluster analysis, which both showed clear regional separation of microbiomes obtained from Austria and Uganda at the species rank. Utilizing PCA analysis, a distinct regional demarcation was evident, particularly at the genus level. However, in Figure S7, this differentiation was not as pronounced among the various sampling locations within Austria (A01 to A05) and a subset of Uganda (U7 to U9) when examined at the family, order, class, and phylum ranks, as demonstrated (Fig. 3).
Fig. 3
Depicting the bacterial species originating from lichen samples gathered in Austria (denoted in red) and Uganda (denoted in green), the PCA plot (a) and hierarchical clustering dendrogram (b) offer visual insights. Furthermore, a set of PCA plots is presented, each corresponding to the genus, family, order, class, and phylum ranks, and these visualizations can be found in Figure S7
Utilizing LDA, the calculation of regional distinctiveness among OTUs was conducted and subsequently marked. This distinctiveness was further identified through LEfSe, and the resultant indicator OTUs or higher taxa were visually represented within the phylogenetic cladogram as illustrated in Fig. 4. Notably, significant indicators, categorized in Table 6, were chosen with the criterion of setting the LDA score threshold to 4. Austria exhibited significant indicators, including OTU KB906754_s (Edaphobacter sp.), Acidisphaera_uc, PAC000374_s (Acidisphaera sp.), AJ292611_s (an unidentified species within the Acetobacteraceae family), GQ495410_s (an unidentified species within the Acetobacteraceae family), HQ622748_s (an unidentified species within the Acetobacteraceae family), LJHX_s (Polymorphobacter sp.), as well as the Acidisphaera genus and AJ292611_g (an unidentified genus within the Acetobacteraceae family), the Sphingomonadaceae family, the Sphingomoadales order, and the Pseudomonadota phylum.
Fig. 4
The LEfSe cladogram (a) and bar plot (b) to visualize taxonomic indicator bacteria that are specific to the regions of Austria (denoted in red) and Uganda (denoted in green) about the collected lichen samples. This cladogram features an innermost node representing the domain of Bacteria, followed by concentric nodes indicating phylum, class, order, family, genus, and species. Nodes or shaded areas in red and green signify significantly higher relative abundances of taxa. The size of each node circle is proportional to the abundance of reads attributed to the corresponding taxon
Table 6 Distinctive indicator OTUs and taxa, characterized by LDA scores exceeding 4, were pinpointed within the assigned OTU diversity of both Austria and UgandaUganda exhibited significant indicators, including OTU HQ622735_s (an unidentified species within the Armatimonadales order), PAC000216_g_uc (an unidentified species within the Armatimonadales order), EU861940_s (an unidentified species within the Acetobacteraceae family), FM874383_s (an unidentified species within the Acetobacteraceae family), PAC000216_g (an unidentified genus within the Armatimonadales order), PAC000074_f (an unidentified family within the Armatimonadales order), as well as the Armatimonadales order, the Armatimonadia class, and the Armatimonadota phylum.
On the rank of species, a total of 11 indicator OTUs (seven from Austria and four from Uganda) were identified with an LDA score > 4, which is shown in Table 6. Reducing the threshold to 3 led to the identification of a combined total of 36 indicator OTUs (14 from Austria and 22 from Uganda) at the species level, which were subsequently employed for the differential abundance analysis using ANCOM-BC. Discerned through the highest LDA scores, have been visualized in Fig. 5, while additional indicators are depicted in Figure S8.
Fig. 5
Significant dissimilarities in relative abundances of specific OTU-derived indicators (with P < 0.05), exhibiting the highest LDA scores, were subjected to ANCOM-BC analysis, discerning between Austria (denoted in red) and Uganda (denoted in green). (a), the most pronounced indicator in Austria, GQ495410_s, belonged to the genus PAC000328_g of the phylum Pseudomonadota. (b), the most pronounced indicator in Uganda, EU861940_s, belonged to the genus PAC000327_g of the phylum Pseudomonadota. Additional noteworthy indicator OTUs are depicted in Figure S8
The metabolic pathways of bacteria associated with lichens in two distinct sampling regions were predicted. Utilizing PICRUSt 2.0, the analysis involved a set of 36 indicator OTUs at the species level, distinguished by LDA scores exceeding 3. Subsequently, the representative sequences of these indicator OTUs were mapped onto KEGG metabolic maps, allowing for the visualization of essential metabolic pathways.
In Level 1 of the KEGG database classification, which represents the hugest metabolic categories within the KEGG database, each indicator OTU was projected onto five major pathways. These encompassed metabolism, genetic information processing, environmental information processing, unclassified, and cellular processes, sorted from largest to smallest, and all organized based on their respective relative abundances. Among these, the "metabolism" pathway showed the highest relative abundances as higher than 50% of OTUs of both sampling regions.
In Level 2, which encompasses sub-metabolic categories within the KEGG database, each indicator OTU was assigned to 25 distinct pathways (as shown in Figure S10). Which In Level 3, representing the finer metabolic categories within the KEGG database, each indicator OTU was associated with a total of 193 pathways (as shown in Figure S11). The most significant difference between Austria and Uganda in level 2 and level 3 metabolic pathways is only 1.0%
In addition, only 11 KEGG Level 3 metabolic pathways, which were selected by the mean proportions’ distances between two regions of >|0.0015| (significant differences at P < 0.05), were found among the indicator OTUs from Austria and Uganda (Fig. 6), suggesting that metabolic pathways predicted from the two regions were relatively similar.
Fig. 6
Snapshot of metabolic pathways on KEGG Level 3 identified within the indicator OTUs from Austria (denoted in red) and Uganda (denoted in green). The horizontal axis represents the average proportions of each pathway, facilitating a comparison between the two regions. Pathway selection was guided by a cut-off criterion of a mean proportion distance greater than |0.0015| between the two regions. Noteworthy distinctions, marked by significant differences at P < 0.05, are indicated on the right side
留言 (0)