
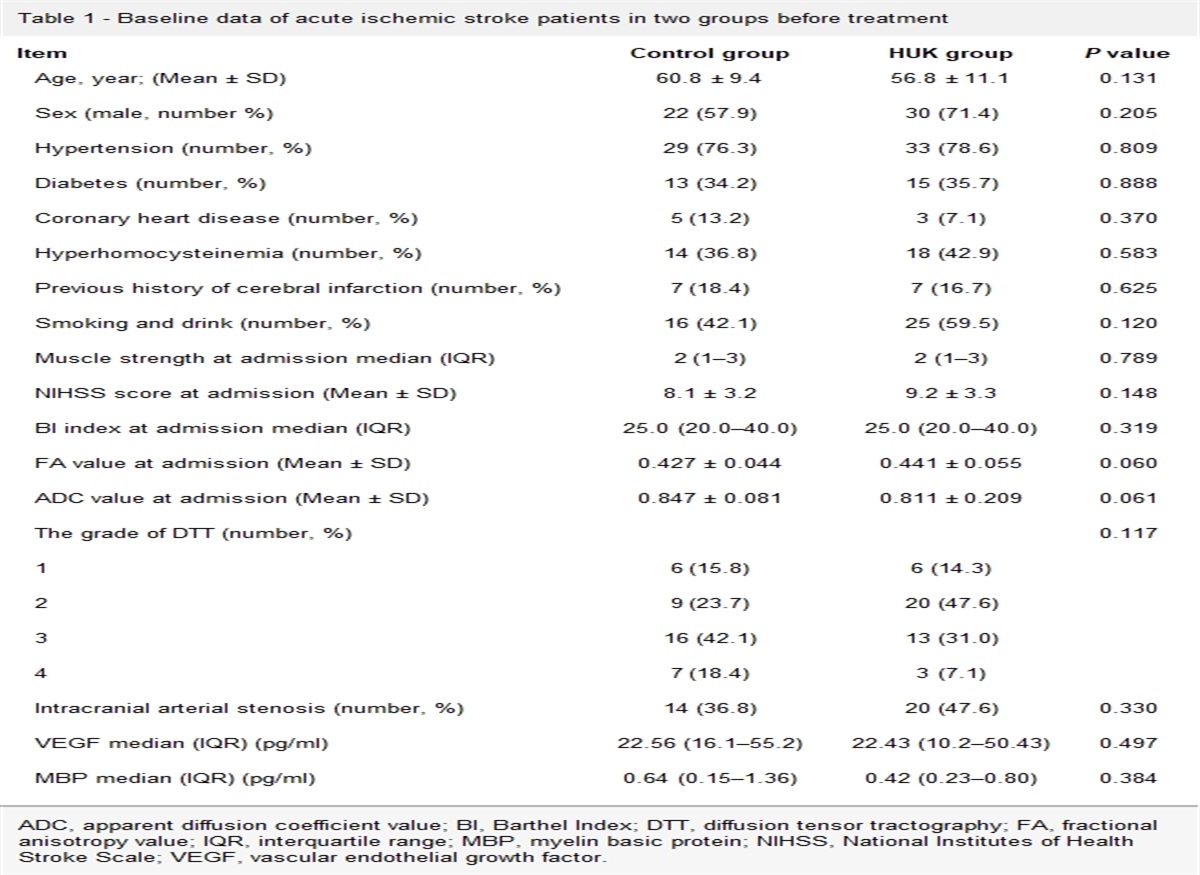
記住我


Brain injury in preterm infants is a leading cause of mortality and disability in children, bringing a heavy economic burden to families and society [1]. Nonetheless, we still lack a deep understanding of its pathogenesis and effective treatment. Recent evidence found that brain injury in preterm infants was a chronic disorder of aberrant brain development. In 2005, Volpe et al. first proposed the concept of ‘encephalopathy of prematurity’, which included the development of abnormalities of white matter and neuron [2]. Then, in 2009, Volpe et al. reported that the abnormalities of brain development and maturation were secondary to primary brain injury, including both white matter (axons and subplate neurons) and gray matter (cerebral cortex, thalamus, and basal ganglia) [3–5]. More recently, in 2021, they reported that neuronal maturation abnormalities were primary, but not secondary to white matter damage [6]. Therefore, aberrant neuronal differentiation and maturation may be the leading cause of brain injury in preterm infants. However, the molecular mechanisms for this remain unclear.
Glycogen synthase kinase-3 (GSK-3) is a conserved serine/threonine kinase and is essential in cell proliferation, apoptosis, and cell cycle [7]. GSK-3β activity is regulated by both of its own phosphorylation sites: Ser9 and Tyr216. GSK-3β-p (Ser9) reduces GSK-3β binding to substrates and inhibits its activity, whereas GSK-3β-p (Tyr216) increases binding and enhances its activity [8]. Recent evidence found that GSK-3β was involved in various nervous system diseases, such as Alzheimer’s disease, schizophrenia, traumatic brain injury, depression, and other cognitive disorders, suggesting that GSK-3β may be a common pathogenic factor for cognitive dysfunction [9].
In 2019, Song et al. found that TWS119, a GSK-3β inhibitor, induced microglial anti-inflammatory activation and improved brain function following experimental stroke [10]. Additionally, our previous study showed that TWS119 reduced neuronal apoptosis and increased synaptic protein expression in hypoxic-ischemic brain damage by activating Wnt signaling pathway and inhibiting the Notch signaling pathway [11]. Thus, Wnt and Notch signaling interactions play a key role in the proliferation and differentiation of neural stem cells [12]. Moreover, one study found that GSK-3β regulated Cyclin D1 expression, a cell cycle regulator [13]. Therefore, whether GSK-3β regulates cell cycle protein expression after hypoxic-ischemic brain damage in immature rats remains unclear.
Cyclins, cyclin-dependent kinases, and cell cycle inhibitors regulate the cell cycle, which was divided into G0, G1, S, G2, and M phases [14]. The G1 phase is the longest and has an important role in the proliferation and differentiation of neural stem cells [15]. Cyclin C, Cyclin D, and Cyclin E, collectively called G1 phase proteins, regulate the G1 phase process. Cyclin D1 positively regulates the cell cycle and p21 negatively regulates the cell cycle [16]. Recent evidence found that cell cycle activation promoted neural stem cell proliferation and inhibited their differentiation into neurons [17]. Therefore, we proposed that the GSK-3β inhibitor, TWS119, promotes neuronal differentiation after hypoxic-ischemic brain damage in neonatal rats by inhibiting cell cycle activation.
This study investigated the effects of GSK-3β inhibitor (TWS119) on cell cycle regulatory proteins, a neuronal differentiation factor (CEND1), maturation neurons, T-box brain transcription factor 1 (TBR1)-positive neurons to clarify the mechanisms of hypoxic-ischemic brain damage in neonatal rats.
Materials and methods Drug administration and experimental designTWS119 (Sigma, SML1271, dissolved in 1% DMSO) was administrated to the 3-day-old Sprague–Dawley (SD) rats by intraperitoneal injection (30 mg/kg) [18]. These rats were randomly divided into 3 groups with random numbers: Sham group: sham-operated rats; HI group: HI and vehicle (1% DMSO) treatment; TWS119 group: HI and TWS119 treatment. At 7 d after HI, These rats were used for investigating the effect of GSK-3β on cell cycle regulatory proteins, neuronal differentiation factor (CEND1), maturation neurons, TBR1-positive neurons. These rats respectively were used for western-blot (n = 8 each group), and for immunofluorescence (IF) (n = 5 each group), and weight of 6.9–10.2g regardless of gender.
HI procedureThree-day-old Sprague–Dawley (SD) rats were used to the hypoxia-ischemic brain damage model, which mimics brain injury of preterm infants [19]. All protocols were made to comply with the National Institutes of Health guide for the care and use. HI procedure was described previously [11]. These animals were anesthetized by enflurane and fixed on the work table; the skin was disinfected with 75% alcohol and cut in the middle of the neck; the left common carotid artery was separated, ligation at both ends and cut at the middle; the skin was sutured and disinfected. The procedure took 3-5 min. These rats were put back to the recovery for 2 h with their mother. Then, these rats were placed in a hypoxic chamber (8% oxygen+92% nitrogen, 37 °C) for 2 h. After HI procedure, these rats were returned to live with their mother.
Tissue processThe neonatal rats were decapitated at 7 d after HI procedure. These neonatal rats were anesthetized and cardiac perfused with 0.9% cold saline and 4% paraformaldehyde. Then, the brains were removed into 4% paraformaldehyde, dehydrated with graded ethanol, vitrified by dimethylbenzene and embedded in paraffin. Next, the embedded tissues were serially sectioned into 5µm coronal sections, which showed cortex, hippocampus and lateral ventricular.
ImmunofluorescenceThese paraffin sections were dewaxed by dimethylbenzene, rehydrated by gradient alcohol, and antigen repaired by citrate. Next, these paraffin sections were incubated with primary antibody: rabbit anti-CEND1 (Abclone, 1:100), rabbit anti-TBR1 (Abclone, 1:100), rabbit anti-NeuN (Abclone, 1:100) overnight at 4 °C. Then, the secondary antibody was 594-conjugated Goat Anti-Rabbit IgG (H + L) (Abclone, 1:100). These sections were stained with 4,6-diamaino-2-phenylinole (DAPI) containing medium. These images were captured with a fluorescence microscope (Leica, Germany). Next, we made the quantification of Neu-N positive neurons. The sections were imaged in DG area with 60× lens and five coronal slices per brain were imaged. The number of cells per field was quantified using Cell Counter plugin for Image J software (National institute of Health, Bethesda, MD, USA). Then we calculated the depth of TBR1-positive neurons in the cortex with the photoshop software (image-analysis-ruler function).
Western-blotThe protein was extracted from the left-brain in ice-cold Radio Immunoprecipitation Assay buffer (Solarbio, China) and the concentration was determined using BCA assay. The samples were separated and transferred to PVDF membranes ((Millipore Corporation, USA). Sequentially, the membranes were blocked with 5%non-fat milk and incubated with primary antibody: rabbit anti-CyclinD1 (1:1000, 55506T, CST), rabbit anti-p21 (1:1000, A1483, Abclone), rabbit anti-GSK-3β (1:1000, 12456, CST), rabbit anti-GSK-3β-p (Ser9) (1:1000, 5558, CST), rabbit anti-CEND1 (1:1000, A5930, Abclone), rabbit anti-TBR1 (1:000, A19550, Abclone), rabbit anti-β-actin (1:5000, AC026, ABclone) overnight at 4 °C. Then, the membranes were incubated in the goat anti-rabbit secondary antibody (1:4000, Abclone) for 1 h. The pictures were taken by The ChemiDocTM XRS + System (Bio-Rad, USA) and the quantitative analysis was performed with the Image J software.
Statistical analysisThe statistical analysis was carried out using the statistical software SPSS20.0. Data was expressed as mean ± SEM. Statistical analysis was conducted using one-way ANOVA. P < 0.05 was considered statistically significant.
Results TWS119 up-regulated the expression of GSK-3β-p (Ser9) after hypoxic-ischemic brain damageFirstly, we investigated the effect of TWS119 on the expression of GSK-3β and GSK-3β-p (Ser9). As shown in Fig. 1, GSK-3β expression had no significant change among Sham group, HI group and TWS119 group (Fig. 1a/c). Whereas, GSK-3β-p (Ser9) expression was significantly decreased in HI group compared with Sham group (P = 0.0072), but significantly up-regulated in TWS119 compared with HI group (P = 0.0036). These results indicated that TWS119 had no significant effect on GSK-3β, but inhibited GSK-3β activity by up-regulating the expression of GSK-3β-p (Ser9)
 Fig. 1:
Fig. 1: TWS119 up-regulates the expression of GSK-3β-p (Ser9) after hypoxic-ischemic brain damage. (a and b) Representative western-blot analysis of GSK-3β and GSK-3β-p (Ser9). (c and d) Quantitative analysis of GSK-3β and GSK-3β-p (Ser9) expression. *P < 0.05, **P < 0.01.
TWS119 inhibited the activation of cell cycle after hypoxic-ischemic brain damageWe demonstrated that TWS119 might inhibit the activity of GSK-3β. Next, we investigated the effect of GSK-3β on cell cycle activation. As shown in Fig. 2, we found that the expression of CyclinD1, a positive regulator of cell cycle, was significantly increased (P = 0.0051), and the expression of p21, a negative regulator of cell cycle, was significantly decreased (P = 0.0469) in HI group compared with Sham group; whereas the expression of CyclinD1 was significantly down-regulated (P = 0.0195), while the expression of p21 was increased (P = 0.0376) in TWS119 group compared with HI group. This suggests that TWS119 inhibits the activation of cell cycle after hypoxic-ischemic brain damage in neonatal rats.
 Fig. 2:
Fig. 2: TWS119 inhibits the activation of cell cycle after hypoxic-ischemic brain damage. (a and b) Representative western-blot analysis of CyclinD1 and p21; (c and d) Quantitative analysis of CyclinD1 and p21 expression. *P < 0.05, **P < 0.01.
TWS119 up-regulated the expression of CEND1 after hypoxic-ischemic brain damageCell cycle exit and neuronal differentiation (CEND1) is an important neuronal differentiation factor. As shown in Fig. 3, we found that CEND1 expression was significantly decreased in HI group compared with Sham group (P = 0.0172), but significantly up-regulated in TWS119 compared with HI group (P = 0.0416) (Fig. 3A/B). By IF, it showed that CEND1 was located in the cytoplasm of mature neurons, and mainly pyramidal neurons in layer III of the cortex. The expression of CEND1 was decreased in HI group, while was increased in TWS119 group (Fig. 3C).
 Fig. 3:
Fig. 3: TWS119 up-regulates the expression of CEND1 after hypoxic-ischemic brain damage. (A and B) Representative western-blot analysis and quantification analysis of CEND1; (C) Representative merged images of the CEND1staining positive cells and DAPT in V2ML of cortex (scale bar = 100 µm), (C - a) Sham group; (C - b) HI group; (C - c) TWS119 group; CEND1 was expressed in the cytoplasm of neurons, which was decreased after HI, and up-regulated in TWS119 group. *P < 0.05, **P < 0.01.
TWS119 promotes neuronal differentiation after hypoxic-ischemic brain damageWe found that TWS119 up-regulated CEND1 expression. Therefore, we investigated the effect of TWS119 on neuronal differentiation by observing the dentate gyrus (DG) area, the main area of neuronal regeneration. We observed fewer neurons in HI group (Fig. 4, A-f) and more neurons in TWS119 group (Fig. 4, A-i). Quantitatively, significantly fewer neurons were present in the DG area (630× magnification) in HI group than in Sham group (P = 0.0007), but significantly more neurons were present in TWS119 group than in HI group (P = 0.0046) (Fig. 4C).
 Fig. 4:
Fig. 4: TWS119 promotes neuronal differentiation after hypoxic-ischemic brain damage. (A) Representative images of the NeuN staining positive cells in DG area, A - a/b/c: Sham group. (A - d/e/f) HI group; (A - g/h/i) TWS119 group; (A - a/d/g) DAPT staining. (A - b/e/h) NeuN staining. (A-c/f/i) the merged images of DAPT and NeuN; (scale bar = 100 µm); (b) the representative images of the merged DAPT and NeuN in DG area (a - Sham group. b - HI group; c - TWS119 group) (scale bar = 30 µm). (C) Quantitative analysis of NeuN-positive neurons in DG area. *P < 0.05, ***P < 0.001.
TWS119 up-regulates TBR1 expression after hypoxic-ischemic brain damageTBR1 is mainly expressed in excitatory neurons and is in layers 5–6 of the cortex. Thus, we examined the effect of TWS119 on TBR1 expression. TBR1 expression was significantly up-regulated in HI group compared to Sham group (P = 0.0061) but significantly down-regulated in TWS119 group compared with HI group (P = 0.0219) (Fig. 5A/B). Next, we analyzed the distribution range and the number of TBR1-positive neurons in the mediolateral area of the secondary visual cortex (V2ML) by IF, finding that the TBR1-positive neurons were primarily located in layers 5–6 of the cortex in Sham group. However, the range of TBR1-positive neurons was significantly larger in HI group than in Sham group (P = 0.0078). Furthermore, TBR1-positive neurons were more concentrated in the TWS119 group than in HI group (P = 0.025).
 Fig. 5:
Fig. 5: TWS119 down-regulates the expression of TBR1 after hypoxic-ischemic brain damage. (a or b) Representative western-blot analysis and quantification analysis of TBR1; (c) Quantitative analysis of the expression range of TBR1-positive neurons in V2ML of the cortex; (d) Representative images of the TBR1 staining positive cells in V2ML of the cortex by IF (scale bar = 100 µm). *P < 0.05, **P < 0.01.
DiscussionGSK-3β is involved in the proliferation and differentiation of neural stem cells, dendrite and axon development, synapses, and neuron transmission [8]. Furthermore, our previous study demonstrated that the GSK-3β inhibitor, TWS119, reduced neuronal apoptosis and up-regulated synaptic protein expression after hypoxic-ischemic brain damage in immature rats via the Notch signaling pathway [11]. The notch signaling pathway has an important role in the brain development [20] and the cell cycle [21]. For example, one study of the myocardium found that Notch2 over-expression increased Cyclin D1 expression [22]. Moreover, Hes1, the target gene of Notch1, inhibited the expression of p21 and promoted the proliferation of neural stem cells [23]. We found that the GSK-3 inhibitor, TWS119, inhibited cell cycle activation by down-regulating Cyclin D1 and up-regulating p21 expression. These results suggest that TWS119 inhibited cell cycle activation, possibly via the Notch signaling pathway.
CEND1, a major regulator of neuronal linear differentiation, is mainly expressed in mature neurons and is a marker protein for neuronal differentiation and maturation [24]. Elesheimer et al. found that the down-regulated CEND1 expression leads to cognitive dysfunction via mitochondrial pathway [25]. Another study also found that the down-regulated CEND1 expression impaired neuronal differentiation, but the up-regulated CEND1 expression ameliorated it [26]. These results suggest that CEND1 is an important regulator of neuronal differentiation and neurodevelopment. Moreover, a previous study of Parkinson’s disease found that the inhibiting GSK-3β promoted neuronal regeneration and inhibited glial regeneration [27]. We found that it decreased CEND1 expression and decreased neuronal differentiation in the DG area after hypoxic-ischemic brain damage. Additionally, CEND1 expression was up-regulated, and more differentiation of neurons occurred in TWS119 group compared to HI group in the DG area. Therefore, TWS119 promoted neuronal differentiation and maturation by the up-regulating CEND1 expression.
Cortex development occurs from inside to outside, first with the inner layers (V–VI), followed by the outer layers (II–IV). TBR1 is a key transcription factor in cerebral cortex development, mainly expressed in Cajal-Retzius cells, subplate neurons, and layer VI glutamatergic neurons [28]. So, the distribution range of TBR1-positive neurons may indicate the neuron migration and cortex development and the number of TBR1-positive neurons may indicates the development of excitatory neurons. A previous study on autism found that the up-regulated TBR1 expression impaired cerebral cortex development and neuronal differentiation [29] However, heterozygous mutants of TBR1 have been found to disrupt cortical development [30]. In our study, TBR1-positive neurons were primarily concentrated in layers (V–VI) and sub-plate neurons of the cortex, and TBR1 expression increased after HI but decreased in TWS119 group, which suggest that TWS119 inhibit the development of excitatory neurons. The increased TBR1 expression comes from the bigger distribution range of TBR1-positive neurons which was involve in neuronal migration and cortical development. These findings suggest TWS119 promote cortex development and inhibit the development of excitatory neurons by down-regulating TBR1 expression.
ConclusionGSK-3β inhibitor TWS119 suppressed the activation of cell cycle, promoted the regeneration of neurons, inhibited the development of excitatory neurons and promoted cortex development after hypoxic-ischemic brain damage. Our findings provide strong evidence for the molecular pathogenesis of brain injury in preterm infants and provide direction for the treatment of brain injury in preterm infants.
AcknowledgementsThis work was supported by the Natural Science Foundation of Hebei Province (No. H2022406039 to Limin Gao) and the Health Commission of Hebei Province (No.20231401 to Limin Gao).
Conflicts of interestThere are no conflicts of interest.
References 1. van Bel F, Vaes J, Groenendaal F. Prevention, reduction and repair of brain injury of the preterm infant. Front Physiol 2019; 10:181. 2. Volpe JJ. Encephalopathy of prematurity includes neuronal abnormalities. Pediatrics 2005; 116:221–225. 3. Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 2009; 8:110–124. 4. Volpe JJ. The encephalopathy of prematurity—brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol 2009; 16:167–178. 5. Volpe JJ. Dysmaturation of premature brain: importance, cellular mechanisms, and potential interventions. Pediatr Neurol 2019; 95:42–66. 6. Volpe JJ. Primary neuronal dysmaturation in preterm brain: important and likely modifiable. J Neonatal Perinatal Med 2021; 14:1–6. 7. Lin J, Song T, Li C, Mao W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res 2020; 1867:118659. 8. Jaworski T, Banach-Kasper E, Gralec K. GSK-3beta at the intersection of neuronal plasticity and neurodegeneration. Neural Plast 2019; 2019:4209475. 9. Manduca JD, Thériault R, Perreault ML. Glycogen synthase kinase-3: the missing link to aberrant circuit function in disorders of cognitive dysfunction? Pharmacol Res 2020; 157:104819. 10. Song D, Zhang X, Chen J, Liu X, Xue J, Zhang L, et al. Wnt canonical pathway activator TWS119 drives microglial anti-inflammatory activation and facilitates neurological recovery following experimental stroke. J Neuroinflammation 2019; 16:256. 11. Gao L, Yang L, Cui H. GSK-3β inhibitor TWS119 alleviates hypoxic-ischemic brain damage via a crosstalk with Wnt and Notch signaling pathways in neonatal rats. Brain Res 2021; 1768:147588. 12. Bejoy J, Bijonowski B, Marzano M, Jeske R, Ma T, Li Y. ‘Wnt-Notch signaling interactions during neural and astroglial patterning of human stem cells’. Tissue Eng Part A 2020; 26:419–431. 13. Takahashi-Yanaga F, Sasaguri T. GSK-3β regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal 2008; 20:581–589. 14. Martínez-Alonso D, Malumbres M. Mammalian cell cycle cyclins. Semin Cell Dev Biol 2020; 107:28–35. 15. Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat Cell Biol 2017; 19:177–188. 16. Coller HA. Regulation of cell cycle entry and exit: a single cell perspective. Compr Physiol 2019; 10:317–344. 17. Bonda DJ, Bajić VP, Spremo Potparevic B, Casadesus G, Zhu X, Smith MA, et al. Review: cell cycle aberrations and neurodegeneration. Neuropathol Appl Neurobiol 2010; 36:157–163. 18. Wang W, Li M, Wang Y, Li Q, Deng G, Wan J, et al. GSK-3beta inhibitor TWS119 attenuates rtPA-induced hemorrhagic transformation and activates the Wnt/beta-catenin signaling pathway after acute ischemic stroke in rats. Mol Neurobiol 2016; 53:7028–7036. 19. Stadlin A, James A, Fiscus R, Wong YF, Rogers M, Haines C. Development of a postnatal 3-day-old rat model of mild hypoxic-ischemic brain injury. Brain Res 2003; 993:101–110. 20. Kim S, Lee M, Choi YK. The role of a neurovascular signaling pathway involving hypoxia-inducible factor and notch in the function of the central nervous system. Biomol Ther (Seoul) 2020; 28:45–57. 21. Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol 2001; 21:5925–5934. 22. Campa VM, Gutiérrez-Lanza R, Cerignoli F, Díaz-Trelles R, Nelson B, Tsuji T, et al. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol 2008; 183:129–141. 23. Kabos P, Kabosova A, Neuman T. Blocking HES1 expression initiates GABAergic differentiation and induces the expression of p21(CIP1/WAF1) in human neural stem cells. J Biol Chem 2002; 277:8763–8766. 24. Wang R, Yang DX, Liu YL, Ding J, Guo Y, Ding WH, et al. Cell cycle exit and neuronal differentiation 1-engineered embryonic neural stem cells enhance neuronal differentiation and neurobehavioral recovery after experimental traumatic brain injury. Neural Regen Res 2022; 17:130–136. 25. Xie W, Guo D, Li J, Yue L, Kang Q, Chen G, et al. CEND1 deficiency induces mitochondrial dysfunction and cognitive impairment in Alzheimer’s disease. Cell Death Differ 2022; 29:2417–2428. 26. Zhou P, Qi Y, Fang X, Yang M, Zheng S, Liao C, et al. Arhgef2 regulates neural differentiation in the cerebral cortex through mRNA m(6) A-methylation of Npdc1 and Cend1. iScience 2021; 24:102645. 27. Singh S, Mishra A, Bharti S, Tiwari V, Singh J, Parul, Shukla S. Glycogen synthase kinase-3beta regulates equilibrium between neurogenesis and gliogenesis in rat model of Parkinson’s disease: a crosstalk with Wnt and notch signaling. Mol Neurobiol 2018; 55:6500–6517. 28. Crespo I, Pignatelli J, Kinare V, Mendez-Gomez HR, Esgleas M, Roman MJ, et al. Tbr1 misexpression alters neuronal development in the cerebral cortex. Mol Neurobiol 2022; 59:5750–5765. 29. Yook C, Kim K, Kim D, Kang H, Kim SG, Kim E, et al. A TBR1-K228E mutation induces tbr1 upregulation, altered cortical distribution of interneurons, increased inhibitory synaptic transmission, and autistic-like behavioral deficits in mice. Front Mol Neurosci 2019; 12:241. 30. Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, et al. Neonatal Tbr1 Dosage Controls Cortical Layer 6 Connectivity. Neuron Cambridge, Mass. 2018, 100(4): 831–845.
留言 (0)