Reagents and antibodies
Roswell Park Memorial Institute (RPMI) 1640 medium, Dulbecco’s Modified Eagle’s Medium (DMEM), L-glutamine, and Antibiotic-Antimycotic (100X) (Anti-Anti) were obtained from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS), phosphate-buffered saline (PBS) and 0.25% trypsin-EDTA were purchased from HyClone (Logan, UT, USA). RES, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), crystal violet, paraformaldehyde, 2′,7′-Dichlorofluorescin diacetate (DCFH2-DA), N-acetyl-L-cysteine (NAC), RNase A, Hoechst 33342 and propidium iodide (PI) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Radioimmunoprecipitation assay (RIPA) lysis buffer and Immobilon Western Chemiluminescent HRP Substrate was purchased from Millipore (Billerica, MA, USA) and protease inhibitor cocktail was purchased from Roche Molecular Biochemicals (Indianapolis, IN, USA). The primary antibody against CD133 (ab19898) was purchased from Abcam (Cambridge, MA, USA). Sox2 (#3579), phospho-Akt (p-Akt (Ser473), #4060), Akt (#9272), phospho-GSK-3β (p-GSK-3β (Ser9), #9332), GSK-3β (#9832), c-Myc (#5605), β-actin (#4970), and the secondary antibody anti-rabbit IgG (#7074) or anti-mouse IgG (#7076) were provided by Cell Signaling Technology (Danvers, MA, USA).
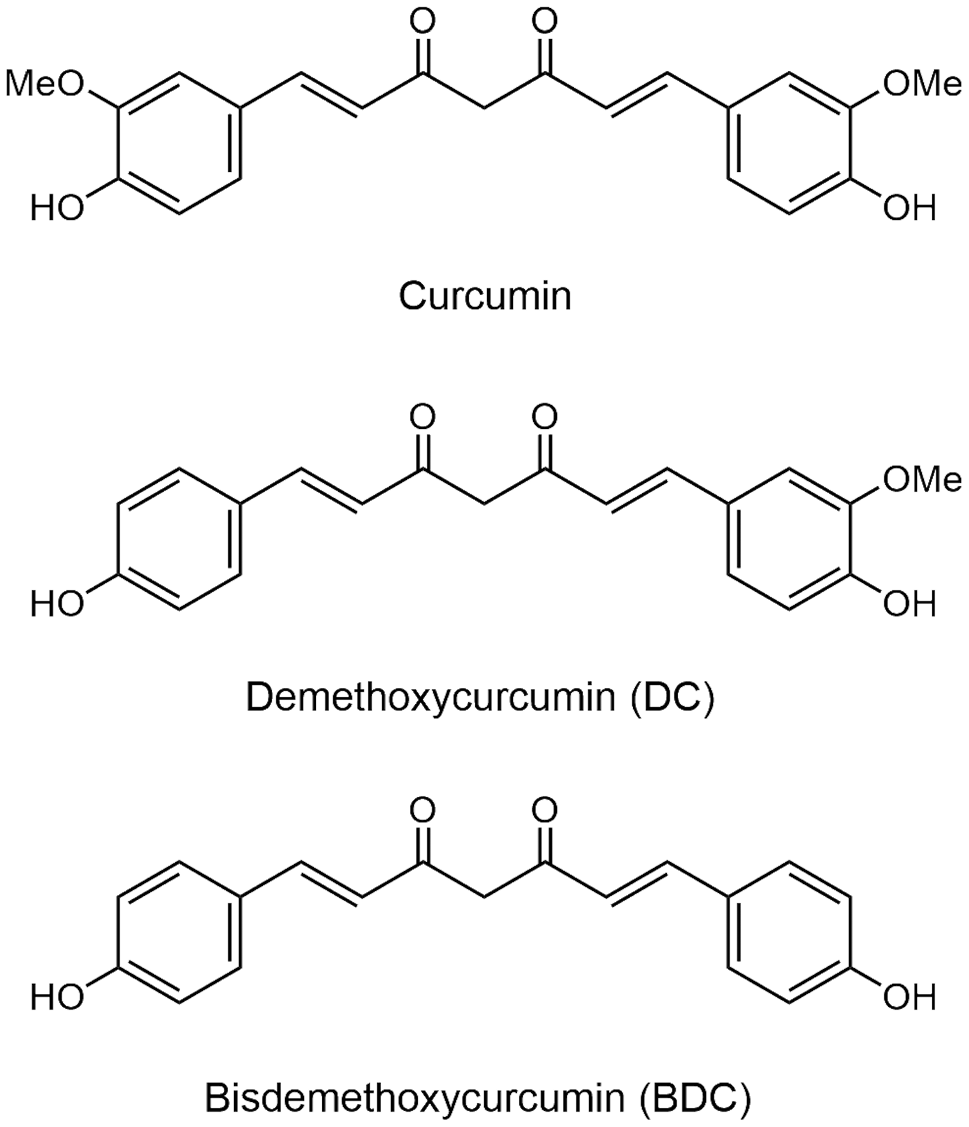
Synthesis of MOS
MOS (SM_7)
A solution of 5 (50.0 mg, 0.104 mmol) in Tetrahydrofuran (THF) (5.6 mL) was hydrogenated over 10% Pd/C (55% water, 22.2 mg) at room temperature (RT) for 20 h. The catalyst was removed by celite filtration and the filtrate was concentrated in vacuo to give MOS (SM_7) (33.6 mg, quant) as a colorless solid.
1 H NMR (400 MHz, CDCl3) δ: 6.84 (1 H, d, J = 7.8 Hz), 6.68 (1 H, dd, J = 1.7, 7.8 Hz), 6.61 (1 H, d, J = 1.7 Hz), 6.34 (2 H, s), 5.49 (1 H, brs), 5.39 (1 H, brs), 3.84 (6 H, s), 3.84 (3 H, s), 2.81 (4 H, s). 13 C NMR (100 MHz, CDCl3) δ: 146.8, 146.2, 143.7, 133.6, 132.8, 132.8, 121.0, 114.1, 111.2, 105.1, 56.2, 55.8, 38.4, 37.9. IR (KBr cm− 1): 3537, 3010, 2939, 1614, 1515, 1463, 1428, 1325, 1220, 1149, 1114, 1034, 927, 770, 662, 566, 557, 541, 518, 512, 500, 486, 457, 532, 426, 420, 412, 407. EI-MS m/z (%): 274 (M+, 32), 138 (11), 137 (100). HRMS (EI): Calcd for C17H29O5, 304.1311; Found: m/z 304.1307.
4-(benzyloxy)-3-methoxybenzaldehyde (1)
A solution vanilline (10.0 g, 65.7 mmol) in CH3CN (54 mL) was added NaHCO3 (6.29 g, 74.9 mmol, 1.14 equiv.) and KI (1.09 g, 6.57 mmol, 0.1 equiv.), and the obtained solution was heated to 60 °C. After benzyl chloride (8.00 mL, 69.5 mmol, 1.06 equiv.) was added to this solution, refluxed for 5 h. After cooling to RT, the reaction mixture was evaporated under vacuum. The residue was diluted with HCl solution (2.1 mL, 1 mol/L) and extracted with EtOAc (50 mL×3), washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified over SiO2 column (n-Hex. : EtOAc = 7 : 3) to give 1 (8.33 g, 52%) as a colorless solid.
1 H NMR (300 MHz, CDCl3) δ: 9.84 (1 H, s), 7.30–7.45 (7 H, m), 6.99 (1 H, d, J = 8.2 Hz), 5.25 (2 H, s), 3.95 (3 H, s).
(4-(benzyloxy)-3-methoxyphenyl)methanol (2)
A solution of 1 (3.00 g, 12.4 mmol) in methanol (30 mL), tetrahydrofuran [(THF) 30 mL], and H2O (3 mL) was added NaBH4 (515 mg, 13.6 mmol, 1.1 equiv.) at 0 °C, and the reaction mixture was stirred for 1 h. The reaction was diluted with Et2O (30 mL) and quenched with HCl solution (11 mL, 1 mol/L). The obtained solution was evaporated under vacuum. The residue was diluted with H2O (10 mL) and extracted with EtOAc (60 mL×3), washed with brine, dried over anhydrous Na2SO4, and concentrated to give 2 (2.99 g, 100%) as a colorless solid.
1 H NMR (400 MHz, CDCl3) δ: 7.43 (2 H, d, J = 7.1 Hz), 7.36 (2 H, t, J = 7.1 Hz), 7.30 (1 H, t, J = 7.1 Hz), 6.95 (1 H, d, J = 1.6 Hz), 6.86 (1 H, d, J = 8.4 Hz), 6.82 (1 H, dd, J = 1.6, 8.4 Hz), 5.16 (2 H, s), 4.61 (2 H, s), 3.91 (3 H, s).
diethyl (4-(benzyloxy)-3-methoxybenzyl)phosphonate (3)
A solution of NBS (7.65 g, 43.0 mmol, 3.5 equiv.) in CH2Cl2 (44 mL) was added dimethylsulfide (3.77 mL, 51.6 mmol, 4.2 equiv.) at 0 °C over 7 min. The reaction mixture was stirred at this temperature for 10 min. A solution of 2 (3.00 g, 12.3 mmol) in CH2Cl2 (44 mL) was cooled at − 18 °C and was added above solution. The reaction mixture was stirred at − 18 °C for 3 h. The reaction mixture was warmed to 0 °C and diluted with H2O and extracted with CH2Cl2 (80 mL×3), washed with saturated NaHCO3 solution and H2O, dried over anhydrous Na2SO4, and concentrated. The crude product was dissolved in triethyl phosphite (2.79 mL, 16.1 mmol, 1.24 equiv.). The reaction mixture was stirred at 140 °C for 4 h. After cooling to RT, the reaction mixture was evaporated under vacuum. The residue was purified over SiO2 column (n-Hex. : EtOAc = 1 : 9) to give 3 (2.01 g, 45%) as a yellow oil.
1 H NMR (400 MHz, CDCl3) δ: 7.29–7.44 (5 H, m), 6.89 (1 H, s), 6.82 (1 H, d, J = 8.0 Hz), 6.75 (1 H, d, J = 8.0 Hz), 5.13 (2 H, s), 3.93–4.11 (4 H, m), 3.89 (3 H, s), 3.04 (2 H, d, J = 21.2 Hz), 1.20 (6 H, t, J = 7.1 Hz).
4-(benzyloxy)-3,5-dimethoxybenzaldehyde (4)
A solution syringaldehyde (10.0 g, 54.9 mmol) in CH3OH (33 mL) was added K2CO3 (9.1 g, 65.9 mmol, 1.2 equiv.) and benzyl bromide (7.82 mL, 65.9 mmol, 1.2 equiv.), and the obtained solution was refluxed for 20 h. After cooling to RT, the reaction mixture was filtered, and the obtained filtrate was evaporated under vacuum. The residue was diluted with H2O and extracted with CHCl3 (50 mL×3), washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified over SiO2 column (n-Hex. : EtOAc = 7 : 3) to give 4 (9.40 g, 63%) as a yellow oil.
1 H NMR (400 MHz, CDCl3) δ: 9.86 (1 H, s), 7.47 (2 H, dd, J = 1.6, 7.3 Hz), 7.28–7.38 (3 H, m), 7.11 (2 H, s), 5.13 (2 H, s), 3.90 (6 H, s).
(E)-2-(benzyloxy)-5-(4-(benzyloxy)-3-methoxystyryl)-1,3-dimethoxybenzene (5)
A solution of 3 (400 mg, 1.10 mmol, 1.2 equiv.) in THF (5.5 mL) was stirred at − 78 °C and added t-BuOK solution in THF (1.46 mL, 1.46 mmol, 1.6 equiv., 1.0 M) over 30 min. The reaction mixture was stirred for 20 min. at the same temperature, and was added 4 (249 mg, 0.915 mmol) in THF (1.0 mL) over 20 min. and the mixture was stirred for 1 h at − 78 °C and for 10 min. at 0 °C. Then, the reaction mixture was stirred for 2 h at RT. The reaction mixture was cooled to 0 °C and diluted with saturated NH4Cl solution and extracted with EtOAc (60 mL×3), washed with saturated NH4Cl solution and H2O, dried over anhydrous Na2SO4, and concentrated. The crude product was purified over SiO2 column (CH2Cl2) to give 5 (231 mg, 52%) as a colorless solid.
1 H NMR (400 MHz, CDCl3) δ: 7.49 (2 H, d, J = 6.8 Hz), 7.44 (2 H, d, J = 7.3 Hz), 7.27–7.39 (6 H, m), 7.07 (1 H, d, J = 2.0 Hz), 6.97 (1 H, dd, J = 2.0, 8.3 Hz), 6.94 (1 H, d, J = 16.1 Hz), 6.88 (1 H, d, J = 16.1 Hz), 6.86 (1 H, d, J = 8.3 Hz), 6.70 (2 H, s), 5.17 (2 H, s), 5.02 (2 H, s), 3.95 (3 H, s), 3.87 (6 H, s). 13 C NMR (100 MHz, CDCl3) δ: 153.6, 149.8, 148.0, 137.8, 137.0, 136.6, 133.3, 130.8, 128.5, 128.5, 128.1, 127.8, 127.8, 127.2, 127.0, 119.6, 114.0, 109.3, 103.4, 75.1, 71.0, 56.1, 56.0. IR (KBr cm− 1): 3547, 3019, 2399, 1507, 1331, 1214, 1030, 928, 753, 668, 501, 476, 454, 441, 435, 429, 407, 401. EI-MS m/z (%): 482 (M+, 13), 392 (28), 391 (100), 91 (62). HRMS (EI): Calcd for C31H30O5, 482.2093; Found: m/z 482.2092.
Cell culture
Human lung cancer H23, H292, and A549 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). H23 and H292 cells were cultured in RPMI medium (Gibco). A549 cells were cultured in DMEM medium (Gibco). The medium was supplemented with 10% FBS (HyClone), 2 mM L-glutamine (Gibco), and 1X Anti-Anti (Gibco). Cells with 70 − 80% confluence were trypsinized with 0.25% trypsin–EDTA (HyClone) and subcultured in the same media. The cells were maintained at 37 °C in an incubator with a humidified atmosphere containing 5% CO2.
Preparation of compounds solution
The stock solutions of RES and MOS was prepared at concentration of 50 and 12.5mM in DMSO (Sigma) and stored in aliquots at − 20 °C. RES and MOS with designated final concentrations (0 − 200 µM) were diluted with cell culture media for subsequence experiments with a maximal DMSO concentration less than 0.5% DMSO. DMSO was used as the vehicle control.
Cell viability assay
The effect of RES and MOS on cell viability in lung cancer cells was assessed by using MTT assay. Briefly, lung cancer cell lines (H23, H292, and A549) were seeded overnight at a density of 1 × 104 cells per well in 96-well plates. The cells were then treated for 24 h with different concentrations (0 − 200 µM) of RES or MOS. After desired incubation, MTT solution (0.5 mg/mL) was added and the cells were incubated in dark for 3 h at 37 °C in the incubator containing 5% CO2. The MTT solution was replaced with DMSO (100 µL/well) to dissolve the purple formazan crystal. The absorbance was measured at 570 nm using a microplate reader (Perkin Elmer, Waltham, MA, USA). Percentage of cell viability in relation to the non-treated control was calculated from the optical density (OD) ratio of treated to non-treated control cells. IC50 values were calculated using regression analysis from dose-response curves (GraphPad Prism7 software, San Diego,USA). The cancer selectivity index (SI) was calculated by the following equation: SI = mean IC50 against normal cells/mean IC50 against cancer cells.
Colony formation assay
Lung cancer cells (H23, A549, and H292) were plated in triplicate into 6-well plates at 300 cells/well. Following overnight attachment, cells were treated with various concentrations (0 − 25 µM) of RES or MOS for 24 h. After treatment, the medium containing RES or MOS was replaced with the fresh medium. The colonies were allowed to form for 7 days, and the medium was changed every two days. For colony staining, colonies were washed once with PBS, then fixed by adding fixative (methanol: acetic acid (3:1, v/v)) for 5 min and stained with crystal violet (0.05% (w/v)) in 4% paraformaldehyde for 30 min. The excess crystal violet was washed with distilled water several times and let air dry at RT. The colonies were photographed with a digital camera, and the acquired images were analyzed using the ImageJ software (National Institutes of Health (NIH), Bethesda, MD, USA).
Cell cycle analysis
The cell cycle distribution was determined by flow cytometry. A549 cells were plated into 6-well plates at 1 × 105 cells/well overnight and synchronized by serum deprivation for 48 h before treatment. The cells were treated with RES or MOS at 0, 1, 2.5, and 5 µM for 24 h. After treatment, cells were stained with PI using the methods previously described [24]. Flow cytometry was performed on a Guava easyCyte HT flow cytometer (Merck Millipore, Billerica, MA, USA). The DNA content in sub-G1, G0/G1, S, and G2/M phases of the cell cycle was assessed using the Guava InCyte software (Merck Millipore).
Measurement of intracellular ROS
The intracellular accumulation of reactive oxygen species (ROS) was assessed by fluorescence microscopy. The lung cancer cell lines (A549 and H23) were seeded in 96-well black plates with clear bottom at 1 × 104 cells/well overnight and treated with various concentrations at 0, 2.5, and 5 µM of RES or MOS for 2 h. After treatment, the cells were stained with 10 µM DCFH2-DA (D6883, Sigma-Aldrich) for 30 min at 37 °C in the dark. Subsequently, stained cells were photographed under a fluorescent microscope (Nikon ECLIPSE Ts2, Tokyo, Japan).
Hoechst 33,342 and PI staining assay
Apoptosis was detected by co-staining with Hoechst 33342 and PI. Nuclear morphology was assessed using the DNA dye Hoechst 33342. The lung cancer cell lines (H23, A549 and H292) were seeded in 96-well plates at 1 × 104 cells/well and treated with various concentrations (0 − 10 µM) of RES or MOS for 24 h. For MOS treatment with NAC, the cells were pretreated with 5 mM NAC for 30 min before being exposed to MOS at 2.5 and 5 µM for 24 h. The cells were stained with Hoechst 33342 (10 µg/mL) and PI (1 µg/mL) for 30 min at 37 °C. Then, the cells were visualized under fluorescent microscope (Nikon ECLIPSE Ts2, Tokyo, Japan) and the percentages of apoptotic cells were determined.
Western blot analysis
A549 and H23 cells were plated overnight. The cells were treated with MOS at 0, 1, 2.5, and 5 µM for 24 h. Following specific treatments, cells were lysed and prepared for Western blotting as was described previously [24]. Nonspecific binding was blocked with 5% skim milk before incubation with the primary antibody (CD133, Sox2, p-Akt, Akt, p-GSK-3β, GSK-3β, c-Myc, and β-actin) at dilution 1:1,000 for overnight at 4 °C. The appropriate secondary antibody (goat anti-rabbit IgG (HRP) or rabbit anti-mouse IgG (HRP)) was diluted at dilution 1:5,000 and incubated for one hour at RT. The enhance chemiluminescence (Immobilon Western HRP Substrate, Millipore) was used to detect protein bands and analyzed by ImageJ (NIH, Bethesda, MD, USA).
Spheroid formation assay
To generate CSC-rich population, A549 cells were grown in a 24-well ultralow attachment plates at a density of 1.5 × 103 cells/well in DMEM medium containing 1% FBS for 7 days to form primary spheroids. At day 7, primary spheroids were harvested and resuspended as single cells using 0.25% trypsin–EDTA (HyClone) and seed onto 24-well ultralow attachment plates for 14 days to form secondary spheroids. After 14 days of secondary spheroid development, each secondary spheroid was collected, dissociated into a single spheroid of the same size, and treated with non-cytotoxic concentrations (0 − 2.5 µM) of RES or MOS for 3 days. Phase-contrast images of secondary spheroids were captured (day 0, 1, and 3) after treatment using a phase-contrast microscope (Nikon ECLIPSE Ts2). At day 3, a single spheroid was co-stained with Hoechst 33342 and PI at 37 °C for 15 min and photographed under a fluorescence microscope (Nikon ECLIPSE Ts2).
Immunofluorescence assay
For staining cell monolayers, A549 and H23 cells were seeded at a density of 1 × 104 cells per well in 96-well plates overnight. The cells were then treated with 5 µM of RES or MOS and incubated for 24 h. Following fixation with 4% paraformaldehyde for 15 min, cells were permeabilized with 0.2% (v/v) Triton X-100 and blocked with 10% (v/v) FBS for 20 min at RT. The cells were incubated overnight at 4 °C in the presence of p-Akt or CD133 primary antibodies at a dilution of 1:100 in 4% FBS. After incubation, Alexa Fluor 488 or Alexa Fluor 594 conjugated with goat anti-rabbit IgG secondary antibody at a ratio of 1:500 in 4% FBS was added and incubated for 1 h at RT in the dark. Cell nuclei were stained with Hoechst 33342 (10 µg/mL) for 15 min at RT and then photographed under a fluorescent microscope (Nikon ECLIPSE Ts2, Tokyo, Japan).
For staining spheroid (3D), A549 cells were allowed to form primary and secondary spheroids as described before. At day 14 of secondary spheroid development, CSC-rich populations were treated with 1 µM of RES or MOS for 24 h, and then the cells were stained following the same procedure as described previously for cell monolayer staining.
Molecular docking
The binding of investigated compounds to Akt1 was performed through molecular docking. The crystal structure of Akt1 (PDB code: 5KCV) bound to miransertib, an oral allosteric Akt inhibitor [25], was downloaded from the Protein Data Bank (PDB). The integrated “loops/refinement” model of UCSF Chimera (version 1.16) [26] was used to reconstruct in the missing residues. The top-ranked model was selected for further analysis. The 3D structures of the natural compounds were downloaded from the PubChem database [27] and optimized with the XTB (version 6.5.0) program package [28] using the GFN2-xTB method with extreme level [29]. UCSF chimera was used to prepare the ligands and receptors, and AutoDock Vina (version 1.2.3) with the Vina forcefield [30] was used to predict the binding modes of compounds. The docking parameters used in this research were the same as in previous reports [31].
Molecular dynamics (MD) simulations and free energy calculations
The predicted binding modes of targeted compounds from molecular docking studies were used as initial structures for the molecular dynamics (MD) simulation using the Amber18 and AmberTools19 packages. The protein and compound parameters were used for the FF14SB force field [32] and generalized AMBER force field version 2 (GAFF2) plus AM1-BCC [33, 34]. All complex systems were immersed in a 10 A° truncated octahedral box of pre-equilibrated TIP3P water molecules, and all system charges were neutralized by adding three chloride counterions. The systems were subjected to energy minimizations using the first 2500 steps of the steepest descent (SD) method followed by 2500 steps of the conjugate gradient techniques in the Sander module. Next, NVT equilibration reached 310 K. All hydrogen bond atoms were constrained by the SHAKE method, and the particle mesh Ewald (PME) algorithm was used to treat long-range electrostatic interactions under periodic boundary conditions. Finally, MD simulations were carried out for 100 ns. The stability between the natural compounds and Akt1 was measured by the root mean square deviation (RMSD) using the module implemented in AMBER 18. The Molecular Mechanics Generalized Born Surface Area (MM/GBSA) method [35] using the MMPBSA.py module in AMBER18 calculated the binding free energy for the complex systems [36]. The UCSF ChimeraX (version 1.4) program was used to visualize 3D molecular structures and interactions [37].
Statistical analysis
All experiments were performed at least three times, and all results are expressed as the mean ± standard deviation. Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA). The unpaired t-test was used for the statistical analyses between two groups. P < 0.05 was considered statistically significant.

留言 (0)