
記住我
Drosophila melanogaster is an established model organism for developmental studies and due to the remarkable conservation of the signaling regulating autophagy, it has been used to better understand the relationship of this catabolic pathway with the genetic conditions that in humans are responsible of a class of neuronal diseases called proteinopathies (PPs). Autophagy is a key cellular pathway that, together with the ubiquitin-proteasome system (UPS), controls protein homeostasis by degrading misfolded proteins or exhausted organelles otherwise detrimental to the cells (Kocaturk and Gozuacik, 2018). Autophagy and UPS are closely linked, in fact protein ubiquitination is a key step for the cargo recognition by the autophagic receptors and alterations in one pathway may affect the activity of the other (Waite et al., 2022). Both pathways are crucial for cell survival particularly in neurons where their perturbation causes age-associated disorders including neurodegenerative diseases (Rai et al., 2022). In this review, we will illustrate the contribution of Drosophila studies to the understanding of the role of autophagy in PPs induced by mutations in genes responsible for the most common neurodegenerative diseases (summarized in Table 1). Furthermore, we will discuss how flies could be used to further improve our understanding of the mechanisms that control these diseases, particularly those that are linked to mutations in genes that are physiologically involved in the control of the autophagic-proteostatic pathway.
Table 1. Drosophila models of human proteinopathies discussed in this article, their principal mechanism, and components of pathways (modifiers) that can either suppress on enhance the toxic phenotype.
2. Drosophila versus human brainDrosophila ‘s studies played a crucial role in understanding brain function and development, leading to significant advances in neuroscience. Drosophila’s brain contains about 200,000 neurons, which are quite similar to mammalian neurons in terms of electrophysiological properties, and form a complex network of interactions recently mapped at high resolution (Zheng et al., 2018), and glial cells that represents 5–10% of the total cell population within the central nervous system (CNS). The CNS is composed of multiple specific and distinct brain compartments (Ito et al., 2014) that are interconnected and synergistically cooperating to control complex behaviors, such as learning, flight control, courtship, grooming, and memory-driven behaviors. Despite the small size, the overall brain organization and regional division shows fundamental similarities to the network structure of the mammalian brain. Indeed, the neural organization underlying primitive functions, such as the perception of odors, taste, vision, sound, gravity, and the circuits regulating feeding and satiety are very similar to those in humans (Tsubouchi et al., 2017; Jayakumar and Hasan, 2018). Drosophila CNS can be divided into two histological regions: the neuronal cell cortex, where all the neurons cell bodies are located, and the neuropil, where axons and dendrites project. This represents a major difference with vertebrates, where the cell bodies are in different areas of the brain depending on the circuit that they are regulating. In addition, in the fruit fly, the lateral connections between neighboring projection-neurons of the same compartment are made not only through interneurons (Liou et al., 2018) but also through dendro-dendritic synapses, which are rare or absent in most vertebrate neuropiles. In terms of morphology, while in vertebrates most neurons are multipolar, in invertebrates there are mostly unipolar (Rolls, 2011; Shah et al., 2016; Smarandache-Wellmann, 2016). Despite these differences, both vertebrate and invertebrate neuronal circuits are plastic which means that their structure and physiology are modified in response to stimuli, both extrinsic and intrinsic, during development and in adult life (Holtmaat and Svoboda, 2009; Lin et al., 2013; Peretti et al., 2015). The electrophysiological properties of the fruit fly neurons are also very similar to those in mammals. Their firing activity depends on Na+ and K+ fluxes that affect the membrane potential, and they communicate using vesicle release of conserved neurotransmitters (i.e., acetylcholine, GABA, glutamate) and neuromodulators (i.e., biogenic amines and neuropeptides) at the synapses. In Drosophila there are several classes of glia, mostly classified based on their morphology function and association with neurons. The perineural glia (PNG), which surrounds the central nervous system, is required to filter nutrients, while the sub-perineural glia (SPG) forms the septate junctions and together form the blood–brain barrier (BBB). This is different than in humans, where the BBB is composed of astrocytes and microglia, however also in Drosophila the BBB prevents paracellular diffusion and controls the influx and efflux of soluble molecules (Hindle and Bainton, 2014; Babatz et al., 2018; Kim et al., 2020). Within the CNS there are other classes of glia cells such as the cortex glia, associated with the neuronal cell bodies, unsheathing glia, that surrounds neuropils and astrocytes that associates closely with the synaptic neuropils and support neurons, similarly to those of the mammalian glia. These classes of glia are abundant and are involved in nervous system development, circuit assembly, synaptic plasticity and neurons support (Freeman, 2015).
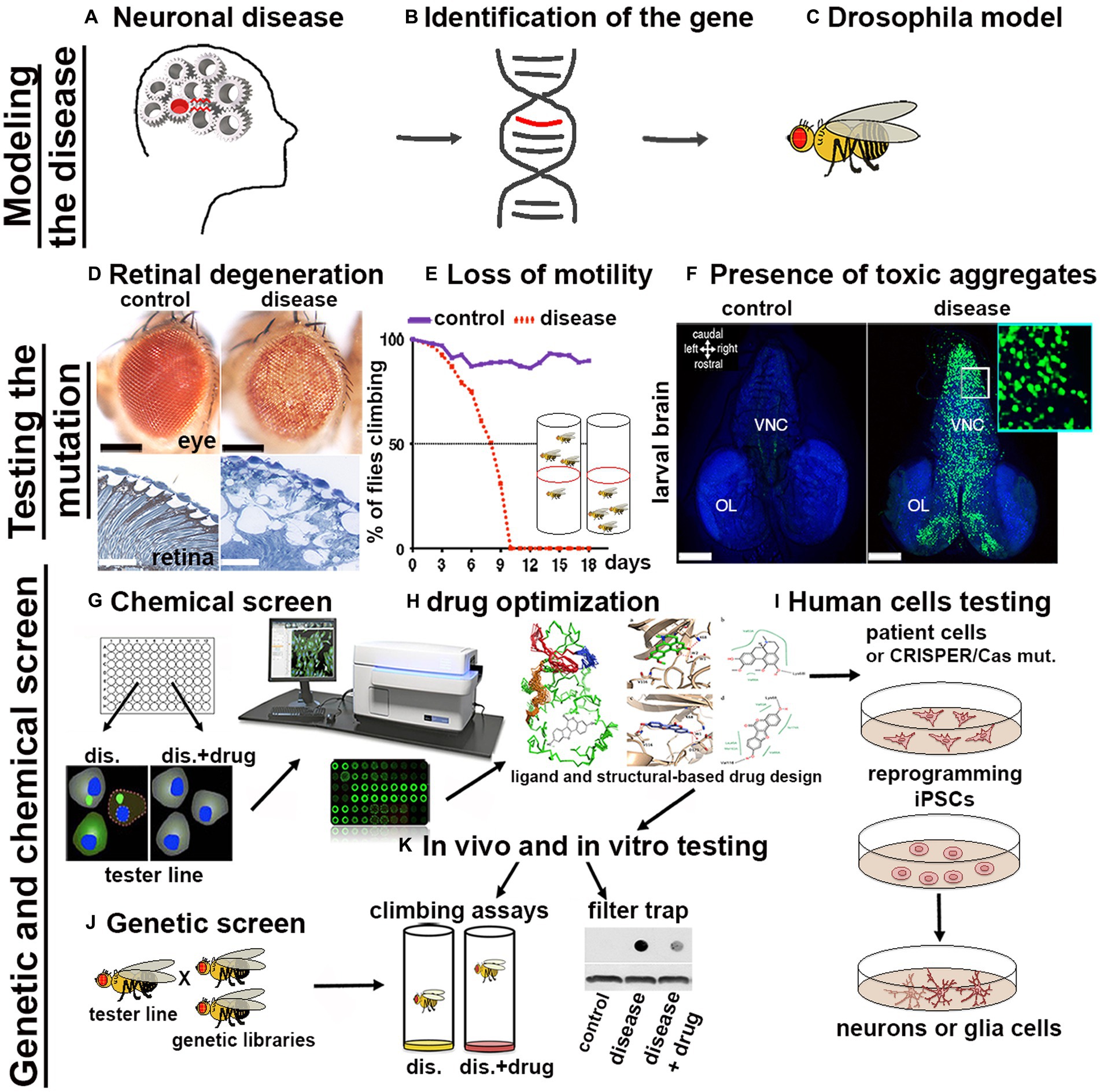
3. Drosophila’s tools to study proteinopathies (PPs)The availability of a wide variety of transgenic strains, advanced genetic tools and databases have enabled the rapid development of Drosophila models for nearly 75% of human neuronal diseases including proteinopathies PPs (Ugur et al., 2016). The simplicity of fly’s brain architecture and the highly conserved function of genes involved in neuronal development, highlights another advantage for its use. Furthermore, Drosophila’s genome generally harbors only one orthologue of the human counterpart therefore, mutation of a single gene generally leads to loss-of-function phenotypes, without redundant effects due to the presence of compensatory paralogues. The availability of unique and advanced genetic techniques allows for the rapid generation of transgenes in which the manipulation of the gene of interest (GOI) is performed in a short time and with unique precision in specific tissues and organs. The most common approach used is based on the yeast derived UAS/Gal4 system, in which a line carrying a tissue specific promoter fused with the transactivating domain of Gal4, is crossed with another line carrying the GOI (overexpression or its RNAi) cloned under the control of the UAS (Upstream regulating sequence). In the progeny, the binding of Gal4 to the UAS sequence will express or reduce the GOI in the tissue of interest (Brand and Perrimon, 1993). If manipulation of the GOI induces lethality, tissue expression control can be achieved by co-expression of the temperature sensitive inhibitor GAL80ts, which acts as a transcriptional repressor of Gal4 at its permissive temperature, or by the Gene-Switch system which relies on a modified version of Gal4 which can be temporally induced by the administration of the drug RU486 (Walters et al., 2019). Variants of the UAS/Gal4 system, such as LexA/Op and QUAS, can be used to drive the expression of different transgenes concurrently with the UAS/Gal4. This is particularly useful for example to study the presence of non-autonomous signals between different organs, tissues or cells in vivo (Potter et al., 2010). Additionally, loss-of-function mutations or overexpression of genes can be easily obtained using CRISPR/Cas9 technology and commercially available sgRNA lines deposited at the Bloomington Stock Center (Ewen-Campen et al., 2017; Zirin et al., 2022). More recently, novel optogenetic techniques have been successfully developed to study protein misfolding in vivo in the brain using Drosophila models of Alzheimer’s disease (AD), Parkinson’s disease (PD), and TDP-43/ALS (Lim et al., 2021). The morphological defects induced by the expression of genes responsible for human PPs (Figures 1A,B) can be easily analyzed after their expression in Drosophila (Figure 1C) using ex vivo dissection of the entire larval or in adult-fly brains, using different techniques ranging from immunofluorescence to super resolution microscopy (SEM or TEM; Figure 1D). Furthermore, motor defects caused by neuronal degeneration can be characterized by analyzing larval motility defects or by analyzing the decline of negative geotaxis in adults, which is the natural ability of flies to climb against gravity (Figure 1E). PPs are characterized by the formation of protein aggregates, which can be visualized by immunofluorescence using specific antibodies, or with the expression of fusion proteins where the gene of interest is fused with fluorochromes such as GFP or RFP (Figure 1F). Biochemical assays, using techniques such as western blot or filter-trap analysis, can show variations in molecular weight and the presence of large insoluble aggregates (Figure 1K). Cell death or induced inflammation at sites of aggregate formation can be studied directly by immunofluorescence assays using appropriate markers (e.g., cleaved caspase, TUNEL assay, or the presence of secreted immune-modulators, like eiger/TNFα or SPARK). Finally, one of the great advantages of using fruit flies as a model for PPs is that the onset of these diseases is relatively rapid, for example the expression of human mutant forms of Huntingtin HTTQ97GFP, in neurons leads to the formation of aggregates which are already visible within 48–72 h of larval development (Figure 1F). This is extremely advantageous since it allows to perform genetic or chemical screening to identify in a short time genes or drugs that reduce the formation of the aggregates (Figures 1G,J). These inhibitors can be chemically optimized (Figure 1H) to improve their qualities and finally be tested on human cells derived from patients (Figure 1I). However, questions remain as to why flies develop aggregates so early in their development, compared to vertebrates. One hypothesis is because flies lack a more complex, adaptive immune system that could control the onset of these diseases as it does in other model systems. In summary, the possibility to target specific mutations into subpopulation of neurons or glia and the ability to rapidly observe their effect cell autonomously or across the neural networks, combined with the short life cycle and the propensity to produce many offspring confirms Drosophila as an excellent model for studying human diseases including proteinopathies.
Figure 1. Scheme of a pipeline to characterize genes associated with proteinopathies and to perform High-throughput Screens with small chemical compounds, to develop new therapeutical strategies in humans. (A–C) From the identification of a gene related to a human disease to the generation of the transformants carrying the GOI. (D–F) The effect of the mutant-disease genes can be tested at cellular and behavioral levels. (D) For example, the expression of exon1 of the human mutant Huntingtin containing 93-CAGs (HTTQ93) in the retina using the GMR-Gal4 promoter leads to loss of the pigmentation in the ommatidia of the compound eye (as seen in the upper image obtained using a stereo microscope) and to retinal degeneration accompanied by defects in tissue morphology and neuronal death [outlined by the white spot of missing tissues visible by transmission electron microscopy (TEM) showed in the images below; (Vernizzi et al, 2020)]. (E) Expression of mutant HTT in neurons using ELAV-Gal4 induces neuronal defects that can be indirectly quantified by measuring the decline over time of the animal motility (negative geotaxis assay). (F) Using the Elav promoter we can express the human HTT exon-1 with 97 CAGs as a fusion protein with GFP (HTT-GFP), and show the formation of HTT-GFP aggregates in larval neurons already at 48-72 hours of age. Photo in panel F, to the right is shown a larval brain of Elav-LexOP-HTTGFP-LexA larvae at 72 hrs AEL, control is to the left. BLUE stains the nuclei (inset 63x). (G–K) Potential pipeline for a High throughput screen (HTS) to identify drugs that reduce the formation of toxic aggregates. (G) Drosophila cells, induced to express the HTTGFP construct are cultured in medium containing small chemical compounds (libraries); analysis of the changes in GFP expression can be quantified using a microplate reader (TECAN). Compounds capable of reducing GFP expression will highlight potential pathways that could be involved in the reduction of mHTT aggregates. (J) Drosophila HD models can be used to perform genetic screens to analyze in vivo the expression of components of these pathways; (K) Drosophila HD models will be fed with the small compounds identified to analyze their effect in ameliorating animal motility or in reducing the size of mHTT aggregates; analyzed directly by immunofluorescence in the brain or by filter-trap assays using either organs or from whole animals. (H) For better performance, chemical drugs can be modified using ligand and structure-based drug design to improve their characteristics and then they can be tested again in vivo in Drosophila HD models such as in K. (I) Finally, the candidate drugs will be tested using human cells differentiated to iPSCs and then to neurons of glia, or directing to neurons iNs (Hong and Do, 2019) starting from cells of patients or from cells from healthy donors in which the specific mutation is introduced using the CRISPR/CAS9 technique.
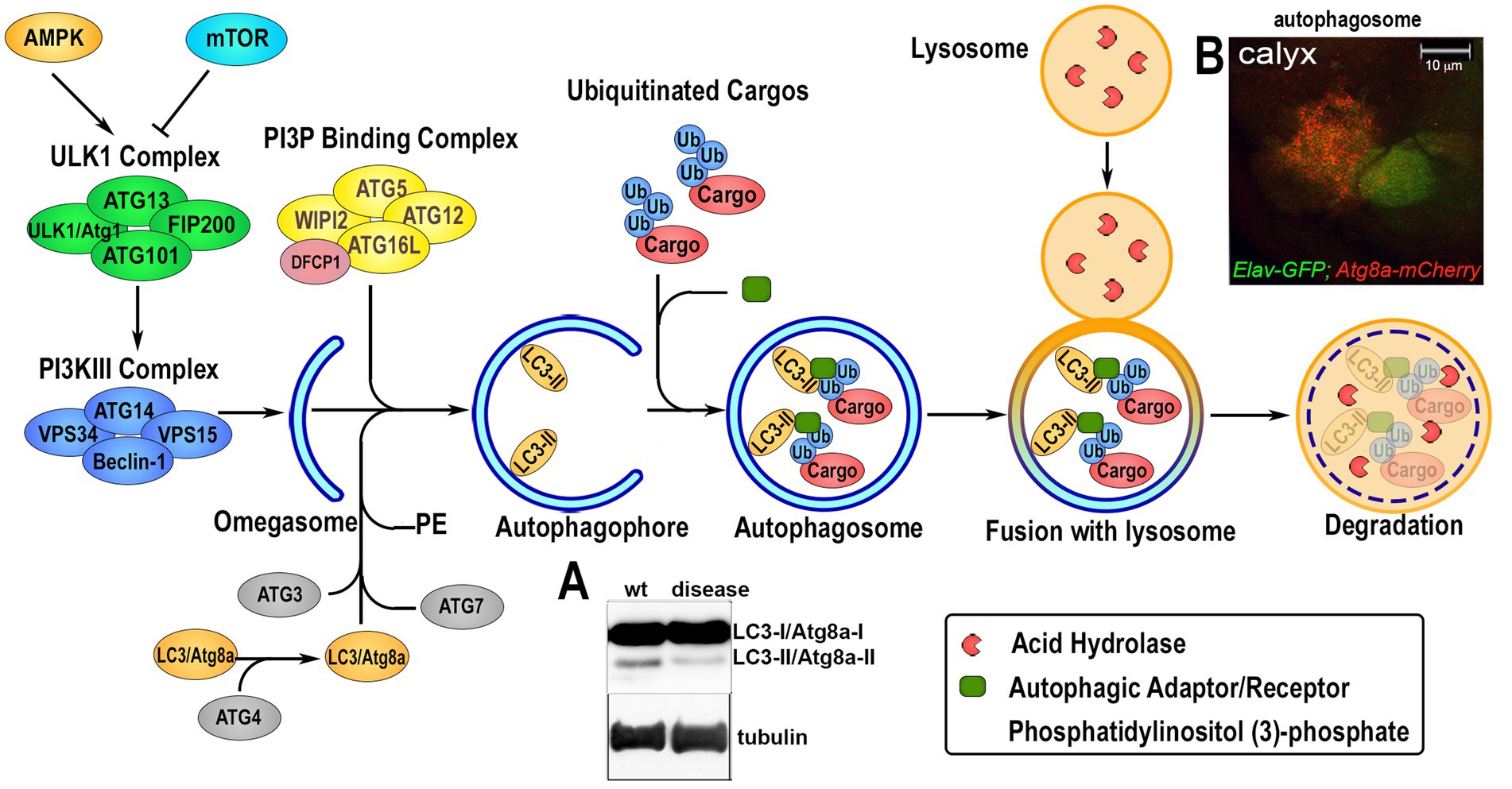
4. Mechanism of autophagyAutophagy is a conserved mechanism with different specific catabolic functions in different tissues and cells. For example, in conditions of nutrient deficiency it represents a survival mechanism that generates amino acids and bioenergetic substrates to allow cell survival. However, autophagy is also used to eliminate toxic debris and exhausted organelles to maintain cellular clearance, particularly relevant in aging neurons, where physiological reduction in autophagy can cause their premature loss. Autophagy can occur in three different forms: microautophagy that is mediated by small cargo-containing vesicles on the lysosomal membrane; chaperone-mediated autophagy (CMA), in which the chaperone Hsc70, a member of the Hsp70 heat shock protein family, recognizes cargo proteins containing KFERQ-like motifs and delivers them to the lysosomes via the receptor lysosome-associated membrane protein 2A (LAMP2A); and the most studied macroautophagy herein referred as autophagy, mediated by a subset of ATG proteins encoded by the ATG gene-family, originally identified in yeast in response to nutrient starvation (Fleming et al., 2022). The product of the ATGs genes is responsible for the initial formation of the phagophore that envelops cytoplasmic cargoes in a double membrane (autophagosomes) which subsequently fuses with the lysosomes for the degradation of the cargo by hydrolases. Due to the specificity of different cargoes, autophagy is now studied and classified into more specific pathways (selective autophagy) such as: lipophagy (lipid droplets), ERphagy (endoplasmic reticulum), mitophagy (mitochondria), pexophagy (peroxisomes), aggrephagy (protein aggregates) and xenophagy (bacteria and viruses; Galluzzi et al., 2017). An important step in selective autophagy is the interaction of the ubiquitinated cargo-proteins with autophagic receptors, such as p62/SQSTM1 (Ref2(P) in flies) that by binding LC3 (ATG8a in flies) via the LIRs (LC3-interacting regions) sequences, recruits cargoes into the autophagosome for degradation. The amino acids produced in the lysosome by hydrolysis of the cargo can directly re-activate the TORC1 complex, located on the lysosomal membrane, thus blocking the autophagic flux (Abu-Remaileh et al., 2017; Wyant et al., 2017). Indeed, TOR kinase, which is part of the TORC1 complex, acts as negative regulator of autophagy by phosphorylating specific sites of the Ser/Tre kinase ULK1/2 (ATG1 in flies) thus destabilizing the initiator complex of the autophagy process that is composed by ATG13, ATG101, FIP200 (Figure 2). This is an essential step for the phagophore assembly since this complex activates local phosphatidylinositol-3-phosphate (PI3P) production at membrane structures called omegasome, that recruits the WIPI2 (WD repeat domain phosphoinositide-interacting proteins) and DFCP1 (zinc-finger FYVE domain-containing protein 1). These proteins are important for the recruitment of the ATG6L1-ATG5-ATG12 complex, responsible for the ATG3-mediated conjugation of LC3 proteins to membrane phosphatidylethanolamines (PEs) to form the membrane double bond. In this process, LC3-I is cleaved at the membrane and converted into LC3-II, process that is also used as a signature of autophagic flux efficiency and typically visualized in western blot assays (Figure 2A). Ectopic expression of LC3 fused to a Fluorescent Proteins is used as a marker of the autophagosome and to quantify the autophagic vesicles (in Figure 2B is shown Atg8a-mCherry expressed in neurons of the calyx together with GFP using the Elav-Gal4 promoter). Autophagy is also regulated by AMPK, a kinase that is activated by cellular stress and low levels of ATP. AMPK phosphorylates ULK1/2 (ATG1 in flies) at sites different than those targets of TORC1. This promotes the formation of the initiator complex of autophagy (Figure 2; Kim et al., 2011). Therefore, molecules capable of inducing basal autophagy, such as rapamycin (a potent TORC1 inhibitor), or metformin (which activates AMPK) represent important therapeutic targets in proteinopathies. The use of simple animal models such as Drosophila was particularly relevant to the discovery and initial characterization of such molecules.
Figure 2. Overview of the autophagic flux. The induction of the autophagic process is regulated upstream by 5†’-AMP-activated Protein Kinase (AMPK) and Target of Rapamycin (TOR), that modulate the activation of the Unc51-like Kinase 1 (ULK1/Atg1) via phosphorylation and the formation of the initiator the ULK1-Complex composed by ULK1, Autophagy-related protein 13 (ATG13), ATG101 and FIP200. Upon activation, ULK1/Atg1 Complex in turn activates via phosphorylation the phosphatidylinositol-3-kinase III (PI3KIII) Complex, which comprises vacuolar protein sorting 34 (VPS34), VPS15, ATG14 and Beclin-1. This allows its translocation on the ER membrane and the production of an isolation membrane enriched in phosphatidylinositol (3)-phosphate (PI3P). PI3P induces the recruitment of the PI3P Binding Complex, consisting of ATG5, ATG12, ATG16L and WD Repeat Domain, Phosphoinositide Interacting 2 protein (WIPI2), on the growing autophagophore (omegasome) and the Double FYVE-containing protein 1 (DFCP1). This process enhances the ATG3-mediated binding of Microtubule-associated protein 1A/1B-light chain 3 (LC3) to phosphatidylethanolamine (PE) on the autophagosomal membrane where is lipidated. This leads to its cleavage from LC3-I/ATG8a-I to LC3-II/ATG8-II, which is considered a feature of an active autophagic flux and can be visualized and quantified by western blot analysis (panel A). Ubiquitin-tagged proteins are recognized by specific autophagic adaptors/receptors, such as p62/SQSTM1/Ref2(P), with a mechanism that is selective for each different organelles or cellular structure called selective autophagy. The cargo receptors bind LC3/ATG8a and transport the cargo into the autophagophore where its content is hydrolyzed upon fusion with lysosome. Autophagosome formation can be visualized in vivo by ectopic expression of LC3/Atg8a fused to GFP or mCherry fluorescent proteins that form small “puncta” on the autophagosome membrane, and fluorescence can be quantified using imaging processing programs. Panel B shows the UAS-Atg8a-mCherry staining pattern under physiological conditions analyzed in the calyx region of Drosophila larval brain, where neuronal cells are visualized by expressing UAS-GFP using the ELAV-Gal4 promoter. Co-localization of mCherry puntae within GFP is used to quantify the presence of the autophagosome in neurons.
4.1. Drosophila’s autophagy a link to neuronal degenerationMost of the steps in the autophagy pathway were first identified and characterized in the Drosophila fat body, where there is a physiological induction of autophagy during metamorphosis to allow the animal to survive starvation (Rusten et al., 2004; Scott et al., 2004). Furthermore, because of the big size of the fat cells (50 μm), the fat body was used to better visualize the autophagosome maturation using ATG8a conjugated with mCherry or GFP (Figure 1B). Subsequently, genetic studies have delineated the important control of autophagy in longevity (Maruzs et al., 2019). First, Atg7d77 mutants showed increased oxidative stress and levels of ubiquitinated proteins, and presented defects in climbing and shortening of lifespan (Juhasz et al., 2007). Similar data were obtained with ATG8a mutants, while its overexpression was shown to promote neuronal survival by controlling oxidative stress and to induce longevity (Simonsen et al., 2008). Work in mice confirmed the role of ATG5 in promoting the survival of Purkinje cells and of ATG7 in preventing axonal degeneration (Komatsu et al., 2007; Nishiyama et al., 2007). Recently, GWAS studies in mice and human cells confirmed ATG5, ATG7 and identified ATG101 and ATG16L1 as crucial for maintaining neuronal survival in particular, ATG7 was associated with complex neurodevelopmental disorders in patients, confirming the role of these autophagic genes in the control of neuronal homeostasis (Wertz et al., 2020; Collier et al., 2021).
5. Neurodegenerative proteinopathies or (PPs)Neurodegenerative Proteinopathies (PPs) are a large family of diseases that share common pathogenic features such as misfolded protein aggregations, neuroinflammation, oxidative stress and mitochondrial dysfunction that contribute to cellular degeneration of the neural tissue (Fleming et al., 2022). Either induced by genetic alteration or by an age-related or stochastic event, alterations of the natural conformation of proteins leads to the formation of oligomers or aggregates with consequent loss of the physiological function of the protein (Rai et al., 2022). Accumulation of aggregates in PPs is commonly due to different mechanisms that include: (i) irreversible formation of aggregates with new strong intermolecular interactions; (ii) inefficacious cell clearance; (iii) seeding and spread of pathological aggregates between cells, evident for prion-like diseases, but identified also in other proteinopathies where the presence of old aggregates function as a seed for the new one (seeds effect; Soto and Pritzkow, 2018). Thus, the central pathogenic role of protein aggregation in these diseases underlines the importance of endogenous mechanisms that control the proteostasis networks, such as the unfolded protein response (UPR), autophagy-lysosome pathway and chaperone activity (Klaips et al., 2018). In fact, age-related disruption of these proteostasis networks or their genetic alterations, contribute to the early onset of these diseases (Fleming et al., 2022; Rai et al., 2022). Deregulation of autophagy has been linked to the pathogenesis on many neuronal diseases thanks to the use of several animal models. In the current review, we will discuss only the most conserved and frequently mutated genes that are the cause of PPs (summarized in Table 1) whose characterization in flies has shown their role in protein homeostasis and autophagy, and those include HD, SCAs, ALS, AD, PD, and prion-like diseases (Pr-D).
5.1. Huntington’s diseaseHuntington’s disease (HD) is an inherited neurodegenerative disorder caused by a dominant mutation in the first exon of the huntingtin (htt) gene that leads to an expansion of the CAG trinucleotide sequence (longer than 35 repeats), resulting in a protein with a long polyglutamine-Q stretch. The clinical onset of the disease is generally at middle age, however the length of the mutation is inversely proportional to the onset of the disease in fact, patients with long CAG show signs already at a young age (Gusella and MacDonald, 1995). The human htt gene encodes for a protein of 350 KDa that contains four HEAT (Huntingtin, elongation factor 3, protein phosphatase 2A and TOR kinase) domains, structurally related to the ARM (armadillo) repeats, and few PEST (peptide sequence rich in proline, glutamic acid, serine, and threonine) domains, that act as substrates for proteolytic enzymes, including caspases or calpain that cleave at amino acids 552 and 586 to produce an N-terminal fragment containing the polyQ domain (Ehrnhoefer et al., 2011). Post translational modifications (PTMs) regulates these proteolytic events and modification of the N-terminus region, such as arginine methylation (Migazzi et al., 2021; Ratovitski et al., 2022), neddylation (Ghosh and Ranjan, 2022), acetylation (Gottlieb et al., 2021), palmitoylation (Lemarie et al., 2021) and phosphorylation at Ser13 and Ser15, are relevant to modulate HTT localization and aggregate formation (Arbez et al., 2017; Chatterjee et al., 2021). These proteolytic cleavages occur also in the mutant HTT (mHTT) resulting in the formation of N-terminus truncated fragments that compete with endogenous HTT and favor the formation of toxic aggregates (Graham et al., 2006; Barbaro et al., 2015; Koyuncu et al., 2017; Ast et al., 2018). The presence of mutant HTT protein induces cellular defects leading to cell death, particularly of neurons in the striatum and cerebral cortex, leading to motor defects and rapid cognitive decline (Bates et al., 2015).
5.1.1. Huntingtin and autophagyThe physical interaction between endogenous HTT and p62/SQSTM1 was shown to facilitates the binding of HTT to ULK1/Atg1 releasing its negative regulation by mTOR, thus promoting autophagy (Rui et al., 2015). The fact that HTT protein contains sequences with structural homology to Atg23, vacuole protein 8 (Vac8ar) and Atg11, containing LC3-interacting repeats (LIRs) sequences (WxxL) present only in cargo receptors suggested a role for endogenous HTT to control autophagy (Ochaba et al., 2014; Martin et al., 2015). Further data showed that myristylation at its N-terminus directed the truncated HTT to the endoplasmic reticulum to initiate autophagosome-formation (Martin et al., 2014). Conversely, the inefficient myristoylation of the mutant HTT promoted the formation of large aggregates that bind p62/SQSTM1 but were not transported to the cargo resulting in an empty autophagosome (Martinez-Vicente et al., 2010; Martin et al., 2015). The human huntingtin gene (htt) is conserved across evolution and its homologue in flies, dhtt, encodes for a protein that shares an overall 24% identity with the human counterpart but contains only one CAG triplet positioned at its N-terminus (Li et al., 1999). While in vertebrates the function of htt is essential for development and is required for neuronal survival (Nguyen et al., 2013), flies dhtt is not necessary for development. However, dhtt knockout animals exhibit a reduced Mushroom body, learning impairment, age-related impaired of motility and shortened lifespan (Zhang et al., 2009). The role of endogenous HTT in autophagy was first outlined in dhtt mutant animals that showed defective developmental autophagy and increased ubiquitination of p62/Ref(2)P (Drosophila’s p62/SQSTM1; Ochaba et al., 2014) and like its orthologue in vertebrates, Ref(2)P accumulated in brains of aged animals (Nezis et al., 2008; Bartlett et al., 2011). Furthermore, endogenous dhtt was shown to counteract the toxic effect of ectopic expression of the mutant human htt-exon-1-Q93 (Zhang et al., 2009), corroborating the protective role of endogenous HTT evidenced also in mice (Van Raamsdonk et al., 2005). More recently, loss of function mutation in the dhtt gene was shown to enhance defects in axon outgrowth in the Mushroom body due to decreased function of the App gene, the Drosophila orthologue of the human amyloid beta-precursor protein (APP) gene, responsible of Alzheimer’s disease, suggesting an interaction between those genes that may control the onset of AD (Marquilly et al., 2021).
5.1.2. Drosophila models of HDA few models of Drosophila HD have been generated over the years (Lewis and Smith, 2016). They are mostly based on the expression of the N-terminal fragments of the human htt gene or on the use of htt-exon-1 mutated to carry different lengths of the CAG tract; while few models have been generated using the full-length mutant human gene due to the high toxicity of its expression when carrying the polyQ expansion tract (Romero et al., 2008). Because of their viability, flies carrying mutations in the htt-exon-1 (mHTT) were widely used to study the function of the polyQ, and to demonstrated that the length of the CAG tract is proportional to a progressive neuronal degeneration and decline in animal motility (Jackson et al., 1998; Marsh et al., 2000; Krench and Littleton, 2017). Furthermore, expression of mHTT in neurons showed a proportional increase in the number and size of aggregates that were already visible at 48–72 h of development (Figure 1F), indicating that Drosophila could be an efficient model to study the kinetic and inhibitors of aggregates formation in vivo. Indeed, genetic screens for aggregate-phenotype modifiers, identified several pathways including TOR signaling (Ravikumar et al., 2004; Sarkar et al., 2007), the chaperons CCT (Sajjad et al., 2014; Pavel et al., 2016), histone deacetylase (Pallos et al., 2008; Jia et al., 2012), antioxidant pathways (Mason et al., 2013), deubiquitinating enzymes (Aron et al., 2018), enzymes involved in glutamine metabolism (Vernizzi et al., 2020), early endosomal protein Rab5 (Ravikumar et al., 2008), Puromycin-sensitive aminopeptidase (PSA; Menzies et al., 2010) and very recently the novel chemical compounds that function as linkers between mHTT and LC3 able to target only the mutant form of HTT to the autophagosome leaving intact the wild-type allele in a selective manner (Li et al., 2019).
5.2. Spinocerebellar ataxiasSpinocerebellar ataxias (SCAs) are a heterogeneous group of inherited disorders that affects the spinal cord and the cerebellum and are characterized by loss of Purkinje cells and cerebellar atrophy. SCAs present mutations in 40 different genes many of which carry an expansion of the polyglutamine tract such as SCA1, SCA2, SCA3/MJD (Machado-Joseph disease), SCA6, SCA7, SCA17 and DRPLA (Dentatorubral-pallidoluysian atrophy; Klockgether et al., 2019). Here we will focus only on the functional mechanisms characterized in Drosophila models for SCA1-3 (Warrick et al., 2005), since models for CACNA1A responsible for SCA6 (Tsou et al., 2016; Sujkowski et al., 2022), SCA7 (Jackson et al., 2005), SCA17 (Ren et al., 2011) and DRPLA (Charroux and Fanto, 2010) only describe motility defect in the adult flies.
5.2.1. SCA1SCA1 is an autosomal dominant disease caused by an expansion of the CAG triplet in the coding region of the Ataxin1 (ATXN1) gene. This gene encodes for a protein with RNA binding capacity that associate to transcriptional regulators at promoter regions (Yue et al., 2001). Patients affected by SCA1 represent 6% of the individuals affected by cerebellar ataxias, and their mutant ATXN1 carries a tract of more than 39 CAG as compared to approximately 20 in the wild-type gene. The mutation leads to the production of a protein with a long polyQ that forms insoluble aggregates, visible in the nuclei of the Purkinje cells (Stoyas and La Spada, 2018). Furthermore, ATXN1 has been shown to favor the toxicity of human pathogenic ATXN2 in mice-models; on the contrary loss of function of ATXN1 increased the stability of BACE1-mRNA, enhancing amyloidogenic cleavage of APP in a mouse model of AD (see Section 5.4.1) and outlining a function of ATXN1 also in AD (Suh et al., 2019).
5.2.1.1. Ataxin1 and autophagyA specific function for Ataxin1 in controlling the autophagic pathway in flies has not been described, however TOR inhibitors in flies were shown to ameliorate the toxic effect of mutant Ataxin1 through the release of the inhibitory role or TORC1 on the lysosomal transcription factor Mitf (Drosophila homologue of human TFEB; Bouche et al., 2016). Drosophila harbors a gene encoding for Ataxin1 (dAtx) that lacks the polyQ domain, nevertheless its overexpression led to phenotypes like those obtained by human ATXN1 overexpression, but different from those observed upon overexpression of the polyQ alone. Genetic experiments showed that Atx1 interacts through its conserved AXH domain with the zinc-finger transcription factor Senseless (Sens). This mechanism is conserved for its mammal’s homolog, the growth factor independence-1 (Gfi-1) and proposed as a potential pathogenic mechanism for SCA1 (Tsuda et al., 2005).
5.2.1.2. Drosophila models of SCA1Expression of the human mutant ATXN1 gene in Drosophila’s neurons leads to retinal degeneration and loss of axonal projections of the interneurons in the ventral nerve cord of the brains (Fernandez-Funez et al., 2000), expression of the pathogenic form of SCA1 or SCA3 in Drosophila larval dendritic neurons reduced their arborization with disruption of F-actin dendritic structures, defect that is partially rescued by Rac-PAK signaling activation (Lee et al., 2011). In vivo screens using the adult eye helped define the role of chaperones to proteins with an expanded polyQ. In particular, the chaperone-dependent E3 ubiquitin (Ub) ligase CHIP (carboxyl terminus of Hsp70-interacting protein) and the NAD synthase nicotinamide mononucleotide adenylyl transferase (NMNAT) together with Hsp70 mediate activation of the proteosome pathway (Zhai et al., 2008). Genetic experiments have delineated the role of transglutaminases (TGs), a class of enzymes that catalyzes the formation of cross-links between glutamine residues within proteins thus increasing their stiffness and insolubility (Muma, 2007). The relevance of these enzymes in proteotoxicity and autophagy was first demonstrated using a Drosophila model for PD and later also in HD, where TG2 phosphorylation by PINK blocked its degradation favoring proteotoxicity and mitochondria degradation. Further experiments, both in Drosophila and in human cells, showed that TG2 reduction ameliorates the defects in both PD and HD (Karpuj et al., 2002; McConoughey et al., 2010; Min et al., 2015). Similarly, a role for TG5 in enhancing the toxicity of mutant ATXN1 was shown in Drosophila models and in cells from SCA1-patients where TG5 was shown to colocalize with Ataxin1 (Lee et al., 2022b) further supporting a functional role for this enzymes in the disease. Genetic screen and chemical inhibitors revealed the RAS-MAPK–MSK1 as an important axis in the control of Ataxin1 aggregate formation (Park et al., 2013). Using small inhibitors of the MSK1/MAPK complex it has been demonstrated in mice and flies that the phosphorylation of Ataxin1 on Ser776 is relevant for its stability, while mutation of this residue in the wt protein reduced the level of Ataxin1-82Q in mice suggesting that Ser776 could represent a novel substrate for both drug and allele-specific therapies (Nitschke et al., 2021).
5.2.2. SCA2Mutations in the human Ataxin2 (ATXN2) gene lead to spinocerebellar ataxia type 2 (SCA2) which accounts for 13% of spinocerebellar ataxias (Klockgether et al., 2019). Mutations in ATXN2 with an expansion greater than 32 CAG repeats forms insoluble cytoplasmatic aggregates visible in Purkinje and granule cells, leading to gliosis and neuronal death (Huynh et al., 2000). Additionally, mutations in ATXN2 were found to be a risk also for ALS due to their effect on increasing TP-43 toxicity (van den Heuvel et al., 2014).
5.2.2.1. Ataxin2 and autophagyNot many studies ruled out the function of autophagy in SCA2 until recently, when it was shown that autophagy ameliorates mitochondrial dysfunction and oxidative stress in mice models for SCA2 and in cells from patients (Wardman et al., 2020). In addition, it was shown that cordycepin, a drug that activates AMPK and autophagy, reduces the abnormal accumulation of p62/SQSTM1 and of LC3 observed in cells form SCA2 patients, indicating a dysfunction in the autophagic flux in the disease (Marcelo et al., 2021). Drosophila possess one orthologue of the ATXN2 gene (Atx2) which encodes for a protein Atx2 that shares 23% of identity and 36% of similarity with the human Ataxin2 protein, but differently Drosophila Atx2 does not contain a long polyQ stretch but three short separate segments of glutamines. Structurally human Ataxin2 contains the Lilke-Sm (Lsm) and Lsm-AD domains found in RNA binding proteins (Satterfield and Pallanck, 2006), and further experiments in human and flies confirm that both Ataxin2 and Atx2 physically bind with the polyribosome suggesting that Ataxin2 may be involved in the control of translation (Satterfield et al., 2002).
5.2.2.2. Drosophila models of SCA2Expression of human mutant ATXN2 in neurons resulted in the formation of aggregates leading to neuronal degeneration that (Lessing and Bonini, 2008). Further work demonstrated that overexpression of Drosophila Atx1 promotes the accumulation of human mutant Ataxin2 aggregates, indicating that the interaction between the two proteins could contribute to the pathogenesis of SCA1 and SCA2. This hypothesis is in line with the idea of a cooperative mechanism between Ataxin1 and Ataxin2, also suggested by the presence of Ataxin2-enriched aggregates in postmortem neurons of patients with SCA1 (Al-Ramahi et al., 2007). Further studies should be conducted using Drosophila’s model of SCA2 to better understand the physiological role of Ataxin2/Atx2 in the control of autophagy and how it could be perturbed in SCA2, to reveal novel mechanisms of intervention.
5.2.3. SCA3Also known as Machado-Joseph disease, SCA3 is the most common type of spinocerebellar ataxia. It is characterized by a progressive neurodegeneration caused by an expansion of CAG at the 3′ end of the coding region of the ATXN3 gene, which from 12 to 40 repeat in the wild-type reaches more than 55 CAGs in the mutant form (Klockgether et al., 2019). The gene encodes for a deubiquitinase (DUB), a class of enzymes that counteracts the action of ubiquitin and important for the control protein stability of TP-43 and HTT (Doss-Pepe et al., 2003; van Well et al., 2019; Tran and Lee, 2022), responsible when mutated of ALS and HD, making DUBs potential targets for proteinopathies.
5.2.3.1. Ataxin3 and autophagyAutophagy is compromised by mutation of ATXN3 since the expression of the autophagic markers p62/SQSTM1 and LC3 was found abnormal in the brain of patients with SCA3. Furthermore, transgenic mice expressing ATXN3-71Q exhibit large aggregates that are reduced in the presence of the autophagy-promoting gene Beclin1 (Nascimento-Ferreira et al., 2011). Puromycin-sensitive aminopeptidase (PSA), a cytosolic enzyme able to digest polyQ sequences (Menzies et al., 2010) was shown to ameliorate also Ataxin3 phenotypes both in flies and mice and to induce autophagy (Menzies et al., 2010). Drosophila, like mammals has two isoforms of Atx3 proteins that contain either two or three ubiquitin interacting motifs (2UIM) with an additional at its C-terminus (3UIM; Johnson et al., 2019). Studies in mammals and flies highlighted the different ability of these isoforms to form aggregates when carrying the polyQ expansion. In particular, 2UIM is more prone to form aggregates than 3UIM, that is rapidly degraded mainly through the proteasome pathway (Harris et al., 2010). Further studies in flies showed that the UIMs motifs of Atx3 interact with the heat shock protein cognate-4 (Hsc70-4) to enhance Ataxin3 mutant aggregation and toxicity (Johnson et al., 2021). This data complement those showing that the auto-protective function of Drosophila endogenous Atx3 depends on its deubiquitinating catalytic activity that indirectly reduce the folding of the toxic polyQ present in mutant Ataxin3 proteins as in others polyQ proteins (Warrick et al., 2005), rather than on the activation of the proteasomal degradation pathway (Tsou et al., 2015).
5.2.3.2. Drosophila models of SCA3The most widely used models for SCA3 express the C-terminal fragment of the human ATXN3 gene with 78CAGs or 82CAGs repeats, that in the retinal neurons cause abnormal eye morphology due to neuronal and tissues degeneration and motility defects (Warrick et al., 1998, 2005). Genetic screens for modifiers of the Ataxin3 mutant phenotypes led to the identification of several genes of the ubiquitin pathways, members of Hsp70/chaperone proteins, and potential regulators of RAN translation, a Repeat-associated non-AUG (RAN) translation that generates toxic dipeptide repeat proteins (DPRs) from pathological repeat RNA expansions hat do not contain the classical methionine start codon (Cleary et al., 2018), see also for C9orf72-ALS (Section 5.3.1). Genetic screen in flies, using the eye, identified members of the ubiquitin ligase family, such as Cullins and of Praja1, that protect against the effect of mutant Ataxin3 strongly supporting the relevance of the ubiquitin and of the DUBs in the disease (Chen et al., 2019; Ghosh et al., 2021). This screens also identified selective modifiers of Ataxin3 able to rescue also Tau-R406W-AD mutation (Section 5.4.2) supporting the existence of common pathways that converge and contribute to neuronal degeneration, controlled by the Ataxin3 DUB-activity (Bilen and Bonini, 2007). In addition, Ataxin3 DUB-activity controls parkin’s ubiquitination in the modulation of “physiological” mitophagy (Durcan and Fon, 2011), and ubiquitination of K63 in the formation of SOD1 aggregates (Wang et al., 2012). Treatment with antioxidant drugs such as the Nrf2-inducers caffeic acid and resveratrol have also been shown to indirectly reduce apoptosis and to induce autophagy, both in human cells and in Drosophila models of SCA3 (Wu et al., 2017).
5.3. Amyotrophic lateral sclerosisAmyotrophic lateral sclerosis (ALS) is a rare disease, with an incidence of 2–3 per 100,000 and a variable progression rate. ALS patients are diagnosed based on the degeneration of motor neurons, however, the pathophysiological heterogeneity of the disease is accompanied by a variety of other factors that are not completely understood. ALS is often associated with frontotemporal dementia (FTD), a disease that presents common mutant genes with ALS resulting in mental disability without motoneurons degeneration or movement impairment. Genetics ALS accounts for only 10–15% of all ALS cases, while the remaining are of sporadic, however up to now, more than 50 genes have been implicated in ALS and the pool of loci associated with this disease is expanding due to GWAS and whole-exome/genome sequencing. Among new pathways identified there are members of sphingolipid signaling and actin polymerization, identified in the vesicles transport network (Brenner and Freischmidt, 2022) suggesting how complex is the physiopathology of this disease. Autophagy in ALS. Deregulation of autophagy has been linked to pathogenesis of ALS thanks to the use of several animal models. ALS patients present accumulation of autophagosomes in the cytoplasm of spinal cord neurons and harbor mutations in genes involved in the autophagy machinery. Many genes have been associated to ALS and here, we will focus on C9orf72, SOD1, TD-P43, FUS since their characterization in flies has been of crucial importance to show their role in protein homeostasis and autophagy.
5.3.1. C9orf72Mutations in the C9orf72 gene are characterized by an expansion of the hexanucleotide sequence GGGGCC (G4C2) in the first intron of the gene, which can reach up to thousands of repeats in the most affected patients (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The gene encodes for a guanine nucleotide exchange involved in cellular processes such as vesicular trafficking, autophagy, lysosomal function and in the control of the immune system and recently described as part of the lysosomal membrane-complex that binds RABs for a correct lysosomal function (Taylor et al., 2016; Root et al., 2021). C9orf72 mutation causes toxicity and alteration of the autophagy-lysosomal pathway contributing to disease pathology (Beckers et al., 2021). Hypothesis regarding the consequences of C9orf72 mutation range from loss of gene function due to epigenetic transcriptional silencing of the locus, to RNA-mediated toxicity caused by the formation of RNA foci that trap both RNA transcripts and RNA binding proteins, and to the formation of toxic dipeptides (DPR) produced by non-AUG translation (RAN) associated with intronic hexanucleotide repeat expansion (Kwon et al., 2014; Wen et al., 2014; Freibaum and Taylor, 2017). The presence of DPRs also compromises the nucleocytoplasmic transport, resulting in the accumulation of toxic debris in the cytoplasm leading to neuronal death (Freibaum et al., 2015; Zhang et al., 2015). Here we have outlined few of these events and discussed their relationship with autophagy.
5.3.1.1. C9orf72 and autophagyThe relationship between C9orf72 and autophagy is somewhat controversial, since some data showed that mutation in C9orf72 cause toxicity and alteration of the autophagy-lysosomal pathway (Beckers et al., 2021), others showed that C9orf72 acts as a negative regulator of autophagy as mice mutant for the C9orf72 gene showed reduction TOR activity with nuclear translocation of TFEB and activation of the autophagy flux (Ugolino et al., 2016). Moreover, DPRs induce dysfunctions of the autophagic-lysosomal pathway by synergizing with other mechanisms of toxicity increasing the pathogenesis of the disease (Beckers et al., 2021). Growth factors and insulin/IGF influences neurodegenerative diseases including AD, PD and ALS. indeed C9orf72-G4C2 mutation has been shown to downregulate insulin/IGF signaling in both fly and human cells while activating insulin/IGF signaling enhances the toxicity of poly-GR repeats (Atilano et al., 2021). IGFs also act as neurotrophic factors for the survival of motor neurons and have therapeutic effects in a mouse model for SOD1-ALS, and in the murine motoneurons NSC34 cells expressing mutant C9orf72-G4C2 (Kaspar et al., 2003; Stopford et al., 2017). Using high-throughput screen to identify chemical modulators of DPRs, we also found that cells expressing mutant C9orf72-G4C2 have increased cAMP levels resulting in protein kinase A (PKA) activation, event that was partially rescued by pharmacological or genetic inhibition of PKA (Licata et al., 2022). Since PKA is activated by growth factors, this observation raises the question of a connection between growth pathways and C9orf72 in ALS and, although the mechanisms are not yet fully understood, we presume that they may converge to the control of cellular proteostasis or autophagy.
5.3.1.2. Drosophila models of C9orf72Although Drosophila does not have a C9orf72 orthologue, transgenic models have been established and mimic the toxicity in humans due to its gain of function effect in flies (Mori et al., 2013; Xu et al., 2013; Mizielinska et al., 2014). The insertion of repeating sequences of GGGGCCs at different lengths was used to determine their different toxicity and ability to form structures called foci that include RNA-binding proteins and RNA (Mori et al., 2013; Mizielinska et al., 2014). RNA of the DPRs obtained by RAN translation was optimized in vitro to express as specific peptides (poly GA, GP, GR, PA, alone or fused with GFP or FLAG tags; Cleary et al., 2018). The difference in toxicity for each DPR is related to their different chemical composition and atomic charge. Indeed, highly arginine-rich GR and PR have been shown to alter the phase separation of LCD-containing proteins by changing the charge and structure of membrane organelles and their dynamics and functions (Wen et al., 2014; Lee et al., 2016). Works in flies evidenced a direct role of C9orf72-G4C2 in autophagy that led to the identification that its expression induced the accumulation of Ref(2)P and of poly-ubiquitinated proteins both in motor neurons and in whole larvae, due to defects in cargo protein degradation. These flies showed defects in lysosome formation, accompanied by reduced Mitf/TFEB nuclear localization (Cunningham et al., 2020). Recent work has demonstrated that nuclear transport of TFEB is mediated by nucleoporins (nuclear import proteins) and their expression is based on the molecular chaperone SIGMAR1/Sigma-1 (sigma-1 intracellular non-opioid receptor) mutated in ALS (Wang et al., 2022). Overexpression of SIGMAR1 in flies restores proper cellular transport, outlining the relevance of this chaperonin in nuclear import in this disease (Lee et al., 2020a).
5.3.2. Superoxide dismutaseSuperoxide dismutase (SOD1) is the second most frequently mutated gene in ALS, representing approximately 20% of familial ALS. This gene encodes for SOD1 a ubiquitously cytosolic Cu/Zn-binding enzyme that homodimerizes and catalyzes the dismutation of superoxide radicals to hydrogen peroxide counteracting the toxic effect of free reactive oxygen species (ROS). SOD1 modulates protein quality control, autophagy, mitochondrial function, and axonal transport. SOD1 mutations may lead to both loss or gain of function phenotypes, making difficult to interpret the mechanisms through which its mutation leads to ALS (Brenner and Freischmidt, 2022).
5.3.2.1. Superoxide dismutase-ALS and autophagywt SOD1 was found in a complex with Atg9/Beclin1 to control P62/SQSTM1 expression, indeed mice mutant for SOD1-ALS show reduced Atg9/Beclin1 and increased level of P62/SQSTM1 and of ubiquitinated proteins (Nassif et al., 2014). Work in Drosophila as well as in C. elegans, identified a role for lethal(2) malignant brain tumor (L3MBTL1) a histone methyl-lysine protein, and SET domain-containing protein-8 (SETD8) in the control protein misfolding and degradation as their reduction alleviate the phenotypes induced by SOD1. This mechanism is conserved also in mammals underlining the role of chromatin modification in the control of protein quality control (Lu et al., 2019). As we know the interplay of ubiquitin-mediated autophagy is controlled also by deubiquitinases (Clague et al., 2019) and studies in Drosophila, as in other invertebrate models, outlined the negative role of the DUB-USP7 that by reverting the ubiquitination of the E3-ligase NEDD4L, reduced autophagy and the clearance of misfolded SOD1 and TDP-43 proteins by reducing the SMAD2/TGF-beta pathway (Zhang et al., 2020).
5.3.2.2. Drosophila models of SOD1Drosophila endogenous dSOD1 is necessary for neuronal health as its null mutation results in neuronal loss. Co-expression of a mutant dSOD1 allele, carrying specific structural LOF mutations, results in formation of unfunctional wt-SOD1/dSOD1 heterodimers indicating that functional dimers are necessary for the activity of the enzyme (Phillips et al., 1995). Studies in Drosophila and in mice demonstrated that expression of human SOD1 in motor neurons reduced ROS activity, leading to longevity (Parkes et al., 1998; Missirlis et al., 2001). Further studies in flies demonstrated that the enzymatic activity of human SOD1 is not required for its toxicity: In fact, homozygous inactive SOD1 mutants (G85R, H48R, and H71Y) expression in dSOD1 null flies results in neurodegeneration, motor defects, and shortened life span, suggesting that these phenotypes are associated to SOD1-mutations rather than SOD1 activity (Şahin et al., 2017; Agudelo et al., 2020). Expression of mutants SOD-1 results in deposition of protein aggregates visible in neurons, glia and in skeletal muscles in both mice-of ALS and cells from ALS patients (Dirren et al., 2015). Mutations in SOD1 lead to mitochondrial impairment, a defect that also occurs in Drosophila, in fact mutant SOD1 accumulates in the intermembrane space of the mitochondrial membranes modifying their structure, reducing the production of ATP and causing metabolic dysfunctions particularly relevant for the activity of motor neurons (Tafuri et al., 2015; Gallart-Palau et al., 2016). A distinctive characteristic of SOD1 mutations is the presence of non-autonomous signals from glia that induce lethality in neighboring cells. This effect was demonstrated in vivo in Drosophila and mice models for SOD1-ALS (Boillee et al., 2006; Watson et al., 2008) and supported by data showing that ablation of the glia ameliorate the disease in mice mutants for SOD1-ALS (Guttenplan et al., 2020). The importance of non-autonomous signaling has been strengthened by recent work demonstrating that motor neurons from SOD1-G93A mutation-bearing mice exhibit metabolic changes that affect surrounding myocytes (Peggion et al., 2022), and likewise between glia and neurons to induce survival of neurons (Chua et al., 2022). Understanding the nature of the non-autonomous signals is relevant to better define the mechanisms in the pathogenesis of ALS and to identify new components and modalities of therapeutic intervention. Thus, the design of Drosophila ALS models using the combination of the two binaries, LexA-LexOp with UAS-Gal4, may allow for the expression of SOD1 mutations in a specific tissue, for example motor neurons, and the concomitant manipulation of glia, using the UAS-Gal4 where many lines are available at stock centers. This would allow to rapidly identify genes that can interfere/suppress SOD1 neuron lethality similar to the approach used for identify mHTT non-autonomous interactors (Bason et al., 2019).
5.3.3. TAR DNA-binding protein 43TAR DNA-binding protein 43 (TDP-43) is a DNA/RNA-binding protein that belongs to the heterogeneous nuclear ribonucleoprotein (hnRNP) family with both nuclear and cytoplasmic functions. Functionally, TDP-43 is involved in modulation of several aspects of RNA life such as transcription, splicing, stability and turnover, degradation, alternative polyadenylation, transport, translation, and microRNA biogenesis [165]. TDP-43 is considered a key protein in ALS for two main reasons: firstly, TDP-43 is the major component of the ubiquitin-positive cytoplasmic inclusions found in spinal motor neurons of ALS patients, secondly, mutations in its gene (TARDBP) occur in around 0.9% of ALS patients.
5.3.3.1. TAR DNA-binding protein 43 and autophagyAutophagy plays an important role in the clearance of the cytoplasmic inclusions in fact, it has been shown that the TDP43 aggregates colocalize with markers of autophagy and inhibition of autophagy increases their aggregates formation (Brady et al., 2011). Moreover, TDP-43 itself modulates autophagy, therefore creating a complex scenario whose perturbation contributes to ALS (Huang et al., 2020). Despite the recognized importance of TDP-43 in ALS, it is still unclear whether the pathogenesis of ALS is related to its reduced physiological function, since wild-type TDP-43 is sequestered in inclusion bodies and unable to interact with its physiological targets, or if it is due to the formation of toxic aggregates containing TDP-43 (Scotter et al., 2015). Loss of TARDBP in mammals downregulates histone deacetylase 6 (HDAC6), an enzyme that controls ubiquitinated protein and autophagy (Fiesel et al., 2010). Controversial results have been obtained in TARDBP knockout cells, where although the lysosomal transcription factor TFEB is upregulated, accumulation of immature autophagic vesicles and reduced expression of Atg7 are observed, suggesting that reduction of TARDBP affects other signals/genes necessary for a functional autophagy (Xia et al., 2016).
5.3.3.2. Drosophila models of TDP-43Drosophila harbors a TARDBP orthologue, namely TBPH, and several groups have investigated its function to gain insights into ALS pathogenesis by performing loss of function studies. Interestingly, Atg7 overexpression suppresses the semi-lethality of TBPH null flies, ameliorating motility, lifespan and reducing the accumulation of Ref2(P)/P62 inclusions at the NMJ (Donde et al., 2020). Other studies in flies addressed the consequences of overexpressing wild-type or disease forms of TDP-43. Overexpression of a cytoplasmic wt-TDP-43 or of TDP-43-M337V mutant in the fly’s retina results in neurodegeneration that can be rescued by co-expression the heat shock protein HSP67Bc that promotes autophagy (Crippa et al., 2016). More recently, HEXA-018, a novel chemical compound that induces autophagy in a TOR-independent manner and ameliorates climbing activity and lifespan was identified as a suppressor of TDP-43 overexpression in flies (Lee et al., 2021).
5.3.4. Fused in sarcomaFused in sarcoma (FUS) is a DNA/RNA-binding p
留言 (0)