
記住我
Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare disease characterized by late-onset asthma, blood and tissue eosinophilia, and small-to-medium vessel vasculitis (1). It was first described in 1951 by two pathologists (J. Churg and L. Strauss), based on an analysis of autopsies of 13 patients with asthma, eosinophilia, and specific organ lesions, such as cardiac insufficiency, renal failure, and peripheral neuropathy (2). Its annual incidence and pre-valence range from 1 to 3 per 1,000,000 and 11 to 45 per 1,000,000, respectively, without gender dominance (3). However, the disease may be underdiagnosed because of restrictive pathomorphological criteria (2). Patients with asthma are a particular risk group, as they experience EGPA 34 times more frequently than those in the general population (4). The mean age at disease onset is approximately 50 years (5), although the disease can also occur in children (6).
Eosinophilic granulomatosis with polyangiitis is often diagnosed in pneumonological departments, where patients are referred due to asthma and lung lesions in chest computed tomography (CT) scans. In a recent study, among 46 consecutive patients hospitalized in a respiratory center because of peripheral eosinophilia and respiratory/lung symptoms (from 2017 to 2019), EGPA was the most common cause of these conditions (45.6%) (7). According to the current nomenclature classification, EGPA belongs to the group of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAVs), along with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) (8), however, it is clearly distinct from GPA to MPA (9, 10). This is a unique disease sharing features of vasculitis and hypereosinophilic syndrome (HES) (11). In addition, these two processes are responsible for the heterogeneous clinical symptoms and phenotypes. Therefore, diagnosis is challenging and requires careful differentiation under mimicking conditions. ANCA are present less frequently than GPA and MPA (up to 30–40% of patients), and primarily target myeloperoxidase (MPO) (9, 10).
Given its rarity and unique features (such as eosinophilia and eosinophilic inflammation), EGPA has often been excluded from AAV studies, which has resulted in a delay in progress in knowledge about the disease compared to other AAVs. However, recently, increasing interest in EGPA as a subject of clinical trials has been observed, and new international projects concerning EGPA are being developed (12). Significant improvements in our understanding of the disease reflect meaningful progress in its early diagnosis and treatment. In this article, we discuss advances in EGPA, including its pathogenesis, diagnosis, and treatment, considering novel drugs that have or are being evaluated to improve patient outcomes.
Eosinophilic granulomatosis with polyangiitis has been defined mainly based on the histologic findings known since the first EGPA description by Churg and Strauss (2). According to the 1994 Chapel Hill Consensus Conference (CHCC), EGPA is defined as an eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, with necrotizing vasculitis affecting small to medium vessels, and is associated with asthma and eosinophilia (13). In 2012, the nomenclature and classification system was revised. The former name “Churg-Strauss syndrome” was replaced with EGPA, and the disease was classified into a new group “ANCA-AAVs” alongside GPA and MPA (8). However, recent data indicate that the current terminology “EGPA” is not entirely appropriate and requires revision. Although it implies that EGPA is a genuine vasculitis (“polyangiitis”), symptoms of vasculitis are not present in all patients, and it is still debated whether patients having asthma, hypereosinophilia, and eosinophil-rich granulomatous inflammation without necrotizing vasculitis, should be determined as having EGPA (14).
2. Pathogenesis and triggering factorsWhile the triggering factors for EGPA remain unknown, our understanding of its pathogenesis has significantly improved. The disease is considered an immune-inflammatory disorder based on the profound immunological dysregulation of both the innate and adaptive immune systems, including T and B lymphocytes, eosinophils, and neutrophils. In addition, genetic pre-dispositions have been reported (15).
2.1. T lymphocytesIn EGPA both Th1 and Th2 pathways are activated, and eosinophils contribute to organ damage (16). EGPA is mainly considered a Th2-response disease. This is evidenced by elevated serum levels of Th2-related cytokines (17, 18) and increased expression of Th2 and regulatory-type transcripts in bronchoalveolar lavage fluid (BALF) cells from patients with active EGPA (19). The T-cell receptors of patients with EGPA show a restricted repertoire (20), suggesting that an antigen-mediated process is likely responsible for their activation (1). Activated Th2 lymphocytes secrete many eosinophilotropic cytokines, including interleukins (IL) 3, 4, 5, 10, and 13, which enhance eosinophil maturation in the bone marrow and their peripheral activation (18, 19). Among these interleukins (ILs), IL-5 is the key cytokine that mediates the release of eosinophils into the bloodstream. It enhances eosinophil production, maturation, and activation and prolongs survival, mainly by inhibiting apoptosis (21), however, it is not responsible for fostering eosinophil infiltration of specific tissues (21). The relevance of the Th2 pathway is underlined by the efficacy of treatment based on the blocking of IL-5. IL-5 receptor (IL-5R) expression is specific to eosinophil differentiation, as it is almost exclusively expressed in eosinophils (22). Targeting IL-5 or IL-5R has become an attractive approach to treating eosinophil-related disorders, including EGPA (23).
Although the Th2 response plays a crucial role, Th17 and Th1 lymphocytes are also involved in EGPA pathogenesis. Th17 cells are specific lymphocytes that produce several proinflammatory cytokines (IL-17A, IL-17F, or IL-22) and are regulated by regulatory T-lymphocytes (Treg), which suppress the immune response and have a protective role in the development of autoimmune disorders (24). Elevated numbers of Th17 cells and decreased frequency of Treg cells have been found in patients with EGPA; the Th17/Treg ratio correlates well with markers of disease activity, and CCR4-active chemokines contribute to eosinophilia (25). The involvement of the Th1 pathway is evidenced by the increased serum concentration of interferon-gamma (IFN-γ) in EGPA patients (26). This cytokine is involved in granuloma formation to protect against the cytotoxic effects of eosinophils. Moreover, Th1 cells were detected in skin lesion biopsies (27), and the gut mucosa of patients with EGPA; the latter has a positive correlation with disease activity (28). Clonally expanded CD8 + T cells have also been described in patients with EGPA, suggesting their pathogenic role in vascular damage (29).
2.2. EosinophilsEvidence supports that eosinophils play a key role in the pathogenesis of EGPA, with abnormal proliferation, impaired apoptosis, and increased tissue toxicity attributed to eosinophil products (5). Their increased number and extracellular protein deposition have been observed in various tissue specimens, including skin (30) and endomyocardial samples (31). The direct toxic effect is associated with the release of cytoplasmic granules upon eosinophil activation (32, 33). However, it can also be an indirect toxic effect as a result of the recruitment and activation of other inflammatory cells (26). There were two types of granule-characterized eosinophils. The primary granule contains Charcot-Leyden crystal proteins and lipid bodies, which are complex inducible organelles that are the site of eicosanoid synthesis, while the secondary granule contains a variety of pre-formed proinflammatory cytokines, enzymes, and growth factors, as well as specific cationic proteins [major basic protein (MBP); eosinophilic cationic protein (ECP); eosinophil peroxidase (EPO); eosinophil-derived neurotoxin (EDN)], which are mainly responsible for specific organ damage (26, 34). The effect of eosinophils depends largely on the tissue involved, however, complications of their accumulation and activation include thrombosis (34, 35), fibrosis (36), and allergic inflammation (26, 34, 37). In addition to being activated, eosinophils secrete many cytokines which enhance the Th2 response, thereby maintaining a vicious circle. Eosinophils are a key source of IL-25. Its elevated concentrations have been found in patients with EGPA and are associated with disease activity and the degree of eosinophilia (38).
In addition to the Th-2 pathway, eotaxins (CCL11-eotaxin, CCL24-eotaxin 2, and CCL26-eotaxin 3) are potent eosinophil activators. They are eosinophil-selected chemokines mainly secreted by endothelial cells but also by T lymphocytes; for example, both IL-4 and 13 released by Th2 cells are synergic promoters of eotaxin synthesis (39). Furthermore, eotaxin 3 is a particularly potent chemoattractant that binds to a specific CCR3 receptor (highly expressed in eosinophils) (22). Increased levels of eotaxin 3 have been described in patients with EGPA and are correlated with disease activity (40, 41).
One case report of Fip1-like1-plateled-derived factor receptor A (FIP1L1-PDGFRA) – positive EGPA implicated the role of tyrosine kinase pathways as drivers for eosinophilia in EGPA (42). The efficacy of imatinib in FIP1L1-PDGFR A-unmutated EGPA has also been previously described (43, 44). These findings indicate a possible shared pathogenic mechanism of EGPA with HES.
2.3. The innate immune systemIncreased IL-33, thymic stromal lymphopoietin (TSLP), and type 2 innate lymphoid cells (ILC2) have been found in patients with active EGPA, indicating that the pathogenesis of EGPA involves interactions between the innate and adaptive immune systems (45). TSLP is a critical mediator of the Th2 response, acting on multiple cell lineages, including eosinophils and ILC2, affecting their maturation, survival, and recruitment. One activator of TSLP is IL-4, which is significantly increased in patients with EGPA. ILC2 are characterized by high expression of transcription factor 3 (GATA3) and production of IL-5 and IL-13 (28), which are key factors involved in the recruitment of eosinophils.
2.4. B lymphocytes, ANCA, and neutrophilsThe role of B lymphocytes in the pathogenesis of EGPA has also recently been highlighted, although not well established, however, the promising results of anti-CD 20 B-cells depleting therapy can support this idea (46). In addition, many patients exhibit an abnormal humoral response, reflecting B lymphocyte activation. Elevated serum concentrations of total immunoglobulin E (IgE) and IgE-containing immune complexes are often observed in patients with EGPA (26). It has also been reported that immunoglobulin G subclass 4 (IgG4) levels are essentially increased (47) and correlated with the number of affected organs and disease severity in EGPA (48). Tsurikisawa et al. (49) showed a significant increase in the proportion of B lymphocytes positive for CD80, CD27, and CD95 in the blood of EGPA patients with frequent relapses, while those with the seldom-relapsing disease had higher CD19-positive B-cell counts and higher serum IgG levels, suggesting that frequently relapsing EGPA is associated with induced B-cell apoptosis. Finally, a comparison of lymphocyte immunophenotypes in EGPA patients showed that, in addition to increased T lymphocyte activity, they correlated with increased plasmablasts and T follicular helper lymphocytes (Tfh), indicating that B-cell activation is involved in the development of EGPA (50).
The presence of ANCA also reflects the activation of B lymphocytes, however, the pathogenic role of these antibodies in EGPA has not been firmly established and is suspected to be similar to MPA. Animal models have shown that MPO-ANCA has a direct damaging effect on endothelial cells, resulting in the development of necrotizing crescentic glomerulonephritis and pulmonary hemorrhage (51). In a human case study, a newborn was reported to develop pulmonary-renal syndrome with the placental transmission of MPO-ANCA (52). A study conducted by Falk et al. (53) made a breakthrough regarding the pathogenic role of ANCA in AAVs. The study proved that ANCA can activate primed neutrophils to produce reactive oxygen species (ROS) and release lytic enzymes that cause necrosis of endothelial cells and adjacent matrix. Unlike GPA and MPA, where the role of ANCA is well established, in EGPA it is still not fully understood. First, ANCA is detected in only one-third of patients, less frequently than GPA and MPA (9, 54, 55). Second, although EGPA and MPA are characterized by the same type of ANCA (anti-MPO), the diseases differ significantly in their clinical phenotype [e.g., renal involvement or diffuse alveolar hemorrhage (DAH) is much more frequent and more severe in MPA than in EGPA] (56). It has been suggested that alternative MPO epitopes, other than those in MPA, develop in ANCA-positive EGPA, contributing to mitigated vascular features (15). Finally, the presence of ANCA in EGPA does not always correlate with symptoms of vasculitis (14). Some authors speculate that for EGPA, a different targeted epitope, a change to the specific epitope conformation, or a failure in the masking process of this epitope by the ceruloplasmin fragment could explain the presence of MPO-ANCA (57).
In recent years, there has been much interest in neutrophil extracellular traps (NETs). NETs are defined as a network of chromatin threads containing histones and proteolytic enzymes (including MPO) that can be released by activated neutrophils to kill bacteria (58). Furthermore, NETs are considered to play an important role in the pathogenesis of AAVs and are a source of ANCA (59, 60). However, a recent study demonstrated enhanced NETs in patients with EGPA with no regard to ANCA status, significantly correlated with blood eosinophil count (61). Eosinophil extracellular traps (EETs) and eosinophil ETosis (EETosis) have also recently been studied in EGPA (62). Mukherjee et al. (63) demonstrated that immunoprecipitated immunoglobulins from ANCA (+) sputum derived from patients with EGPA allowed extensive EETs from both neutrophils and eosinophils in vitro. Direct evidence of EETs/EEtosis within the thrombus in patients with EGPA has been also provided (64).
2.5. GeneticsSeveral immunogenetic factors that pre-dispose patients to EGPA have been identified. It has been shown that the HLA-DRB1*07 and DRB1*04 alleles are associated with the development of EGPA, while DRB1*03 and DRB1*13 are protective (65). Another genetic risk factor is HLA-DRB4, which suggests a strong link with CD4 + T lymphocyte activation (66). In turn, functionally relevant variations in the IL-10 gene promoter (IL-10.2 haplotype) are associated with ANCA-negative EGPA (67).
Recently, a genome-wide association study (GWAS) demonstrated that ANCA status in EGPA is associated with a specific genetic background (56). EGPA with ANCA positivity is associated with human leukocyte antigen DQ (HLA-DQ), which shares both clinical and major histocompatibility complex (MHC) associations with anti-MPO AAV. In turn, ANCA-negative EGPA has a mucosal barrier origin and is associated with variants of the glycoprotein A33 (GPA33) and IL-5/interferon regulatory factor 1 (IRF1) (genotype sharing with asthma). There was an association of both EGPA subgroups (ANCA + and ANCA –) with variants at the TSLP, BCL2L11, and CDK6 loci and suggestive evidence for BACH2, Chromosome 10, and lipoma preferred partner (LPP), indicating that EGPA is characterized by certain genetic variants associated with the syndrome as a whole (56).
3. Triggering factorsThere are no well-known triggering factors of EGPA, however, environmental factors, infections, and drugs have been speculated. Several cases of disease development following massive antigen inhalation (grain dust, flour dust, and cereal dust) (68) and exposure to pigeons have been described (69). Regarding infectious agents, Aspergillus fumigatus triggers EGPA. Some reports demonstrated that Aspergillus might be a pathogen common to both allergic bronchopulmonary aspergillosis (ABPA) and EGPA, and prolonged exposure to this fungus in some patients with ABPA may promote progression to EGPA (70). A case of concomitant ABPA and EGPA after Aspergillus niger infection has also been reported (71). Other infectious agents include viruses, among others. A case of EGPA following COVID-19 has been recently reported (72).
Other factors include drugs mainly used in asthma, such as leukotriene receptor antagonists (LTRAs) or anti-IgE antibodies, which are also suspected to induce EGPA (73, 74), however, the mechanism to induce vasculitis is not well-known. One hypothesis is that the administration of these drugs in the asthmatic phase of undiagnosed patients with EGPA may result in vasculitis burst due to reducing the steroid dose, previously masking symptoms of EGPA (5). Two case-controlled studies concluded that treatment with LTRAs did not increase the risk of EGPA (4, 75). However, a recent monocentric retrospective study found a significant correlation between LTRAs exposure and ANCA positivity in EGPA patients. The authors speculated that LTRAs could induce imbalanced stimulation of leukotriene receptors, which may cause neutrophil activation, NETs production, and subsequent ANCA stimulation, resulting in the development of vasculitis (76). Other suspected drugs include anti-IL therapies. Ikeda et al. (77) described a case of EGPA that became apparent following the discontinuation of dupilumab (anti-IL-4/IL-13 antibody). Additionally, Lim et al. (78) reported a case of EGPA during benralizumab (anti-IL5Rα) treatment.
As asthma is a major feature, allergy may also contribute to the development of EGPA. However, systematic allergy testing in patients with EGPA revealed evidence of allergy in less than one-third of patients (79). Other suspected factors include vaccination and desensitization (5). A case of EGPA that developed following a booster dose of the anti-SARS-CoV-2 vaccine has also been reported (80).
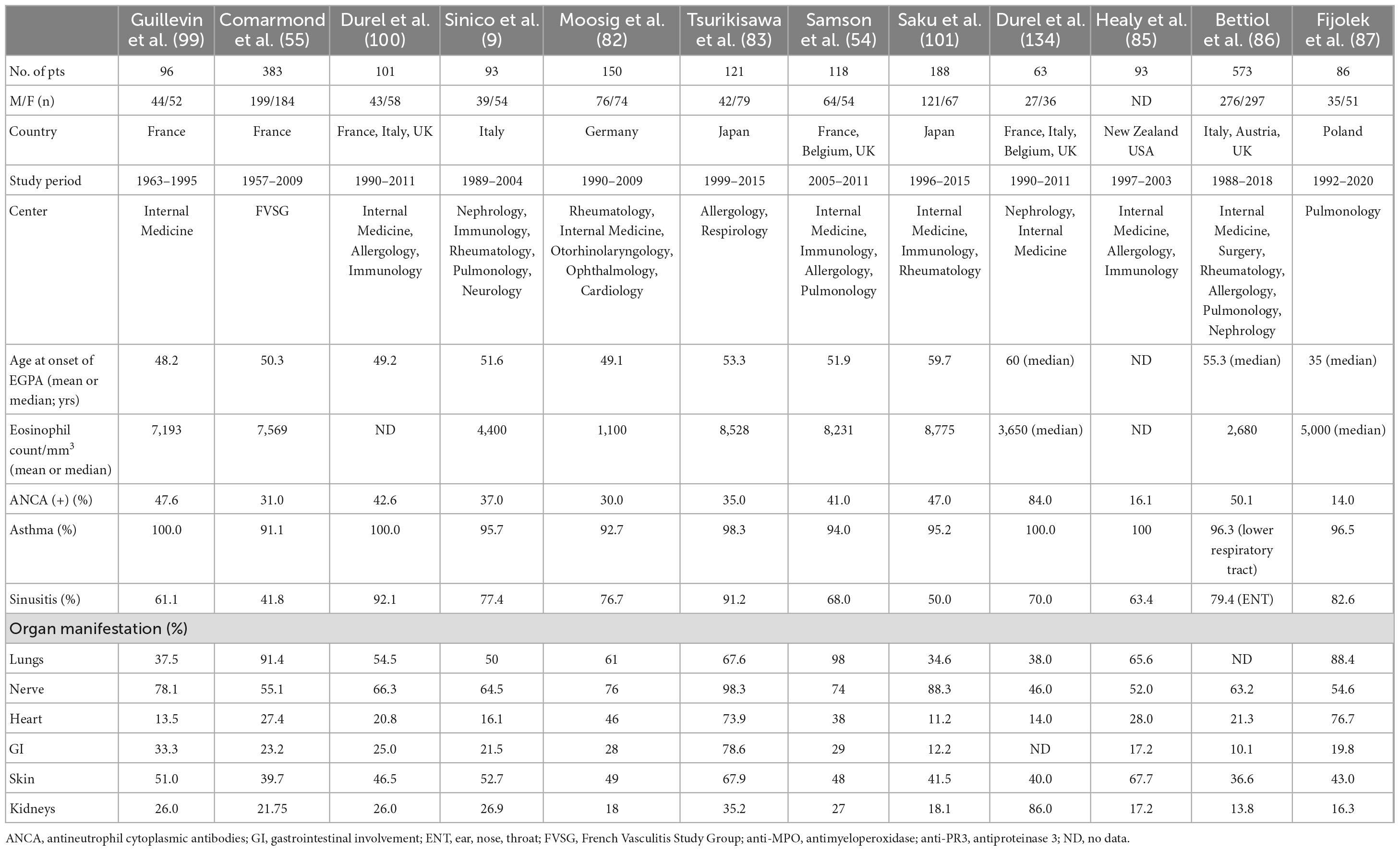
4. Clinical symptoms and disease stagesClassically, EGPA develops in three consecutive stages. The first is the prodromal phase dominated by asthma and allergic rhinosinusitis. After a variable period (mean 9.3 ± 10.8 years) (5), the eosinophilic phase develops—characterized by peripheral and tissue eosinophilia, which may result in pulmonary infiltrates, eosinophilic cardiomyopathy, or gastrointestinal involvement (GI). Next, the disease progresses into the vasculitic phase, in which organ manifestations consistent with vasculitis pre-dominate (81). However, disease succession does not always occur. In some patients, there is an overlap of these phases, or the disease may begin with the eosinophilic phase; in others, the absence of either eosinophilic or vasculitic phases is observed (1, 34). This complexity of the disease makes the clinical manifestation diverse. Interestingly, the spectrum of manifestations varies depending on the patient recruitment center, e.g., patients admitted to respiratory departments have more frequent cardiac involvement and limited features of vasculitis (14). The frequencies of organ involvement and phenotypic features in the selected EGPA cohort are presented in Table 1.
Table 1. Organ involvement and phenotypic features of selected eosinophilic granulomatosis with polyangiitis (EGPA) cohorts.
4.1. The prodromal phaseAsthma is a major feature of EGPA usually preceding the symptoms of vasculitis (mean 9.3 ± 10.8 years) (5). It concerns 90–100% of patients (14, 54, 82–87) and is characterized by distinct features compared to asthmatic patients in the general population. First, it is usually late-onset asthma, which begins in adulthood at around 30–40 years of age. Second, an allergic background is present in less than one-third of patients with EGPA, compared with approximately 70% of patients with asthma in general, and there are no seasonal exacerbations (79). Atopy, if present, is associated with a better prognosis but with more severe or uncontrolled asthma manifestations in the year before the development of vasculitis (88). Third, asthma in EGPA is usually severe and often requires long-term treatment with oral corticosteroids (CS) despite the regression of systemic disease. In a retrospective study of 157 patients with EGPA, asthma was severe in 57% of cases, whereas persistent airflow obstruction was present in 38, 30, and 46% of patients at diagnosis, 3-year follow-up, and final visit, respectively (89). In another study, airflow obstruction was observed in approximately 40% of patients in clinical remission (90). It remains unclear why systemic therapy controls systemic manifestations in EGPA, but not asthma symptoms. Some authors speculate a dissociation between eosinophil bone marrow production and eosinophil recruitments in the airways which results that in sputum (but not blood), eosinophilia is still present in the group of EGPA patients in remission phase (91). Asthma, although often severe, may paradoxically improve during the full-blown vasculitic phase (92). However, it has recently been demonstrated that the severity of asthma increases 3–6 months before the onset of systemic symptoms (89). Furthermore, severe or uncontrolled asthma is associated with baseline pulmonary and ear, nose, and throat (ENT) manifestations but not with clear-cut vasculitic features (93).
Finally, asthma in EGPA is often accompanied by allergic manifestations in the upper respiratory tract, such as allergic rhinitis, chronic sinusitis (70–90%) (89), and nasal polyps (42–58%) (89, 94–96). At this stage of the disease, distinguishing prodromal ENT symptoms in the course of EGPA from chronic rhinosinusitis with nasal polyps (CRSwNP) is challenging; especially in the biopsy, both typical histological features of eosinophilic polyposis are present (96, 97). Lesions observed in GPA, such as destructive granulomatous inflammation or nasal crusting, are uncommon in EGPA. However, secretive otitis media, chronic ear drainage, sensorineural hearing loss, and facial nerve paralysis may occur (34, 98).
4.2. The eosinophilic phaseIn this phase, clinical symptoms are due to eosinophilic infiltration of organs. Typically, the lungs, gastrointestinal tract, and heart are affected.
Lung involvement is present in 37–98% of patients with EGPA, depending on the study series (9, 55, 84, 99–102). In addition, a chest radiograph is abnormal in 70% of patients and shows bilateral pulmonary consolidative or reticulonodular opacities in a peripheral distribution (103). In high-resolution computed tomography (HRCT), which is a more precise method, pulmonary lesions can be classified as airspace and airway patterns (104), however, both types often coexist in one patient. Furthermore, all lung imaging changes observed in EGPA are not EGPA-specific and are frequently observed in other diseases (7, 105). The airspace pattern is mostly migrating patchy infiltrates with peripheral dominance corresponding to chronic eosinophilic pneumonia (EP), (104, 106) which antedate systemic vasculitis in 40% of cases (81). Other common findings are ground-glass opacities (39–53%), followed by consolidations (28–42%), and poorly defined nodules (24–63%) (89, 106). The airway pattern consists of small centrilobular nodules, tree-in-bud sign, bronchial dilatation, wall thickening, and mosaic perfusion pattern (89, 104, 106), which reflect airway involvement in the course of asthma generally, not only in EGPA (7). Greater severity and longer duration of asthma (>5 years) are significantly associated with a higher incidence of airway abnormalities on HRCT in patients with EGPA (107). Histologically, small nodules correspond to eosinophilic bronchiolitis and peribronchiolar vasculitis, whereas bronchial wall thickening is associated with airway wall eosinophil and lymphocyte infiltrations (106).
Other less frequent thoracic symptoms of EGPA include pleural effusion and hilar or mediastinal lymphadenopathy (108, 109). Pleural effusion may develop secondary to eosinophilic pleurisy as well as eosinophilic cardiomyopathy-associated congestive heart failure (1). Other HRCT findings may include interstitial edema, cardiac enlargement, or pericardial effusion, all of which are related to cardiac involvement. In some patients, these HRCT findings may be the only chest symptoms.
A small proportion of patients (3–4%) may experience DAH, which is a life-threatening vasculitic manifestation that can lead to acute respiratory distress (16).
GI is less common in EGPA, although it is significantly more frequent than in GPA or MPA (84). This organ manifestation is recognized in 24–78% of patients, depending on the series and diagnostic tests used (54, 55, 82–84, 100). Manifestations are non-specific and include abdominal pain, which is the most frequently reported symptom (30–91%) (54, 100, 110), followed by diarrhea (45%) (110) and minor bleeding (3–9%) (54, 82, 100). Cholecystitis, pancreatitis, intestinal infarction, and ischemic colitis have been described, but they are rarely present (1–3%) (54, 102). In a study of 383 patients with EGPA, symptoms of acute surgical abdomen occurred in approximately 6% of the cases (55). In another study, 22–45% experienced severe GI manifestations, potentially requiring surgery (16). In EGPA, clinical GI symptoms and findings on abdominal CT are non-specific and require differentiation from other diseases. Common CT features include bowel enlargement and pathologic enhancement (16), whereas histological examination demonstrates mainly eosinophilic infiltrations, sometimes with vasculitis and eosinophilic granulomas (5, 102, 111).
Among the three types of AAVs, cardiac involvement (CI) is most common in EGPA and is mostly present in ANCA-negative patients (9, 10, 55, 112). In Churg and Straus’s original cohort, it occurred in more than 50% of autopsies (2), however, its reported incidence varies from 11 to 74%, depending on the series and diagnostic techniques used (54, 55, 82, 84, 101, 113, 114). Clinical manifestations are variable and include myocarditis (often with thrombus formation), pericarditis, valvular insufficiency, or involvement of the conduction system, resulting in arrhythmia (5, 98, 115–117). The severity of clinical symptoms varies from mild to clinically overt and life-threatening. Patients most often complain of chest pain and dyspnea (116, 118, 119), but the first symptom may also be acute congestive heart failure, life-threatening arrhythmia, and cardiac death (119). In addition, cardiac involvement can be asymptomatic (83, 118, 120–123). In recent data of Polish 86 patients with EGPA, cardiac invasion was found in 76.7% of the cases, with almost 30% of the cases being asymptomatic (87).
Eosinophilia and its cytotoxicity play a crucial role in heart damage caused by EGPA (119). Patients with CI have been reported to have significantly higher eosinophil counts at diagnosis than those without this organ manifestation (118, 124); usually, they were younger, had negative ANCA, higher disease activity, and higher C-reactive protein (CRP) levels (118). Three successive stages of eosinophilic cardiac damage have been described. The first stage is necrosis due to the infiltration of eosinophils and the release of granular proteins. The second phase is characterized by thrombosis formation, whereas fibrosis of the endocardium and valves occurs in the final stage, resulting in restrictive cardiomyopathy and cardiac insufficiency (119). This phase corresponds to scarring of the endomyocardium and is irreversible; therefore, early detection of cardiac involvement is crucial for prognosis. This is because treatment at the earlier stages provides a chance to reverse the inflammatory process and limit myocardial necrosis.
CI of EGPA can also be derived from coronary vasculitis, which is a rare situation occurring in approximately 3% of patients and manifests as myocardial infarction with negative results on coronary angiography (82, 113).
4.3. The vasculitic phaseThis phase manifests as a feature of vasculitis. Typically, the nervous system, skin, and kidneys are affected, with the latter being the rarest. However, every organ may be involved. This phase is often preceded by general symptoms such as fever, weakness, muscle pain, or arthritis.
Involvement of the nervous system is a prominent feature of the vasculitic phase. It affects 42–76% of EGPA patients (54, 55, 82, 84, 87, 102), mainly ANCA-positive (9, 10). Among other forms of AAVs, it is most prevalent in EGPA (65 vs. 23% in MPA, and 19% in GPA) (125). Frequently affected nerves include the peroneal, tibial, ulnar, and median nerves, but the typical presentation is mononeuritis multiplex, usually manifested by foot drop and symmetrical polyneuropathy, often progressing when left untreated (126). Patients complain of numbness, burning sensation, pain, limb weakness, and other sensory disturbances, which can be the first symptom, even in 63% of the cases (126, 127). Diagnosis is mainly based on clinical evaluation and may be confirmed by electromyography (EMG) or nerve biopsy. However, the latter procedure is infrequently performed in clinical practice. In a large study of 955 AAV patients, only 12% underwent nerve biopsies, of which 53% had definitive vasculitis (125). Pathophysiologically, nerve damage is caused by vasculitis and eosinophilic infiltrates, with the latter pre-dominating in ANCA-negative cases (128).
Central nervous system (CNS) involvement in EGPA is less common and is reported in 5–29% of cases with neurological symptoms (54, 55, 82, 83). The main neurological manifestations included ischemic cerebrovascular lesions (52%), intracerebral and/or subarachnoid hemorrhage (24%), loss of visual acuity (33%), and cranial nerve palsies (21%). The clinical course varies, with long-term neurological sequelae being common (43%). Intracerebral hemorrhages have the worst prognostic impact (129).
Skin involvement is the next most prominent feature of the vascular phase. Its frequency ranges from 23 to 68% in patients (54, 55, 83, 84, 87, 100, 101, 113), with vascular purpura being the most common (24–39%) (54, 55, 100, 113). Other findings include subcutaneous nodules that occur in 30% of cases (5) and less frequently, non-specific maculopapular rash, urticaria, petechiae, sterile pustules, livedo reticularis, vesicles, and pruritus (130). A wide range of histological changes is observed in the purpura of the skin, from eosinophilic vasculitis to leukocytoclastic vasculitis without eosinophilic infiltration, making diagnosis difficult (131). Other skin lesions in EGPA show extensive infiltration of eosinophils and surrounding inflamed small dermal blood vessels (132), however, eosinophil infiltration is not specific to EGPA and is a common finding in a broad spectrum of skin diseases (133).
In EGPA, renal involvement is less frequent and less severe than in other forms of AAV (134). In addition, its reported frequency depends on the profile of the medical facility. According to various studies from different centers, the frequency varies from 16.3 to 35% of patients (54, 55, 82–84, 87, 100, 113), with nephrological facilities even in 86% of patients presenting with renal diseases at vasculitis diagnosis (134). Renal involvement in EGPA pre-dominates in ANCA-positive patients, which is in line with the aforementioned study, in which 84% of patients had a positive ANCA test (134). The most common clinical symptom reported in different series was proteinuria (3.3–20%) (55, 82, 113), with renal insufficiency observed in 4.3–15% of cases (55, 83), and up to 75% of patients referred to nephrological facilities, in whom acute renal failure was the most common renal presentation (134). Histologically, the most typical pattern included pauci-immune necrotizing glomerulonephritis (78%), followed by membranous nephropathy (10%) and membranoproliferative glomerulonephritis (3%), both of which were ANCA-negative. Other findings include pure acute interstitial nephritis (10%) and interstitial eosinophilic inflammation in half of the patients, regardless of ANCA status (134).
5. Diagnosis, classification, and disease phenotypesThe diagnosis of EGPA is challenging and requires the correlation of clinical, laboratory, radiologic, and histopathologic findings, however, in cases with a history of asthma, eosinophilia, and both “vasculitic” and “eosinophilic” organ damage, the suspicion of EGPA is quite straightforward - in contrast to those with incomplete manifestations, which can be difficult to recognize. In addition, some patients lack evidence of vasculitis or ANCA, and there is an ongoing debate over whether EGPA can be recognized in these cases. Histology can confirm the diagnosis of EGPA, but the simultaneous presence of all three typical lesions is rare (135). In clinical practice, the diagnosis of EGPA is mainly clinical, however, considering the rarity of the disease and the variety of symptoms, the accuracy of the diagnosis increases with a multidisciplinary discussion among experienced clinicians (16, 103).
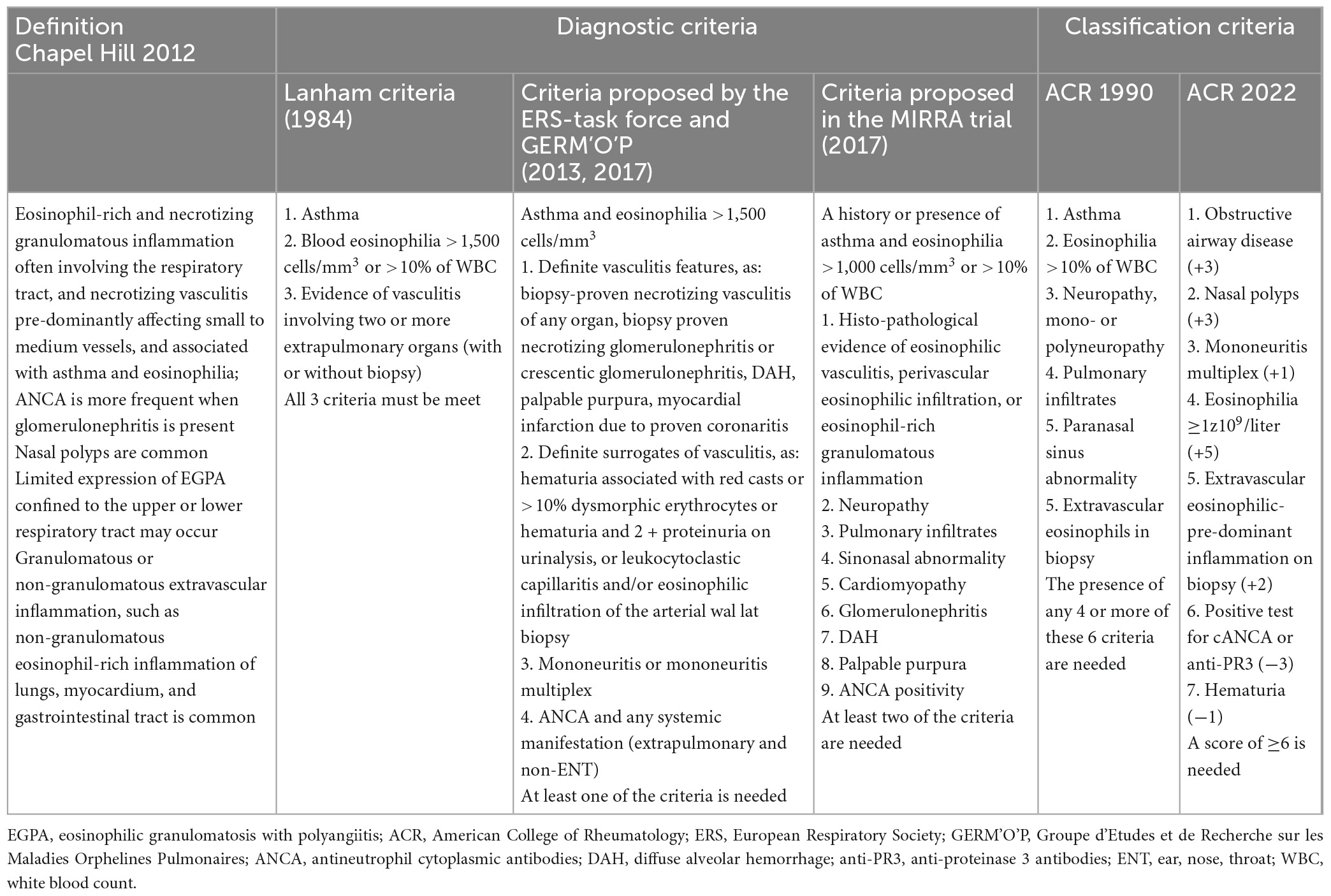
5.1. Diagnostic and classification criteriaTo date, there are no validated or universally accepted diagnostic criteria for EGPA. The aforementioned CHCC is a nomenclature classification and not a diagnostic classification (8, 13). The first diagnostic criteria were proposed by Lanham et al. (81) which included asthma, eosinophilia ≥ 1,500 cells/μL, and manifestations of vasculitis involving at least ≥2 extrapulmonary organs. These criteria were developed before classifying EGPA into AAVs and do not require histological examination. However, they have been widely used by clinicians owing to their simplicity in capturing the essence of the disease. Recently, the Joint Task Force of the European Respiratory Society (ERS) and the Foundation for the Development of Internal Medicine in Europe (Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires; GERM’O’P) proposed new diagnostic criteria (14, 136). They restricted the EGPA terminology to ANCA-positive cases and/or to those with genuine features of vasculitis (or with surrogates of vasculitis) that are precisely defined. In addition, they proposed that patients with asthma, blood eosinophilia, and systemic manifestations, but non-vasculitic and without ANCA, are referred to as having hypereosinophilic asthma with systemic manifestations (HASM), not EGPA. The next criteria are those used in the MIRRA study assessing the safety and efficacy of mepolizumab in patients with EGPA (23). In contrast to the above-mentioned criteria, they were very loose, with the majority of patients not having ANCA or features of vasculitis. However, these criteria were developed for the purposes of a clinical trial (as an eligibility criteria), and are not widely used in clinical practice.
Classification criteria are often mistakenly used as diagnostic criteria, although they are not. Classification criteria were designed to distinguish EGPA from other types of vasculitis; therefore, they should be used only when a diagnosis of small- or medium-sized vessel vasculitis has been established.
The first classification criteria for EGPA were published in 1990 by the American College of Rheumatology (ACR). They were developed by comparing 20 EGPA-diagnosed patients with 787 control patients with other forms of vasculitis and included six items: asthma, eosinophilia > 10%, neuropathy, pulmonary infiltrates, sinusitis, and extravascular eosinophils in the biopsy. The presence of ≥4 of these six criteria allowed the classification of vasculitis as EGPA (137). These criteria were characterized by low sensitivity (67.1%, with 17% of cases meeting the criteria for other vasculitides), and although the specificity was high (64–98.9%), up to 27% of the comparators fulfilled at least one of these criteria (12). Despite poor methodology and lack of validation, these criteria have remained unchanged for several decades. In 2022, the ACR/EAAR (European Alliance of Associations for Rheumatology) established new classification criteria based on a prospective international multisite observational study (Diagnostic and Classification Criteria in Vasculitis; DCVAS project) conducted at 136 sites from 32 countries, including 107 cases of EGPA and 450 comparators. These criteria highlight the significance of peripheral eosinophilia, asthma, and eosinophilic inflammation and specify other features that function as important disease classifiers (such as mononeuritis multiplex, obstructive airway disease, or nasal polyps). Moreover, unlike the previous 1990 criteria, these are validated, have excellent sensitivity (85%) and specificity (99%), and incorporate ANCA testing. The criteria include seven items that have been assigned a point weight (positive or negative), and vasculitis could be classified as EGPA if the cumulative score was ≥6 points (138). Although these criteria were developed primarily for clinical trial purposes, they represent a major advancement in clinical practice as well, however, they are only for EGPA classification and do not solve the problem with diagnosis. A summary of the proposed diagnostic and classification criteria for EGPA (including the definition of the disease) is presented in Table 2.
Table 2. Eosinophilic granulomatosis with polyangiitis – definition, diagnosis and classification.
It is worth noting that owing to its dual nature, EGPA has been also listed as an “associated syndrome” in the classification of HESs (11).
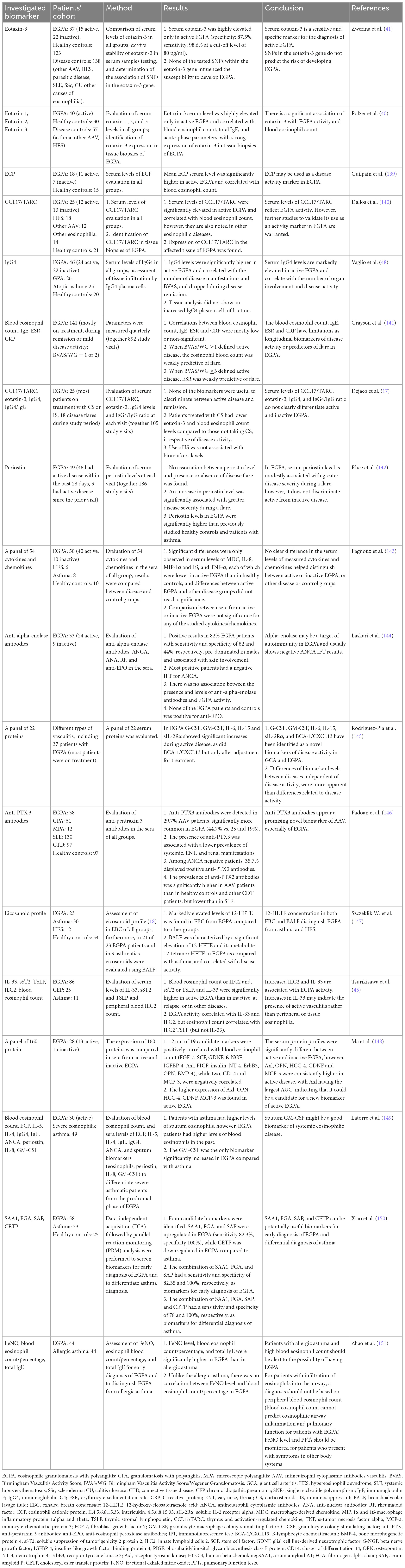
5.2. Diagnostic tests and differential diagnosesTo date, there are no reliable biomarkers of EGPA. The results of these studies were inconclusive, with varying success rates (Table 3) (17, 40, 45, 139–151). Active EGPA is characterized by marked eosinophilia, usually ≥1,500 cells/μL or >10%, which correlates with disease activity (1, 5). It is a fixed feature of EGPA and an important diagnostic criterion, however, in patients treated with systemic CS (e.g., asthma), eosinophil count may rapidly decline within a few days, and the results may be falsely normal (5). A significant proportion of patients have elevated inflammatory markers, such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), mainly at the onset of the disease (55). Non-specific elevations in IgE levels were detected in 75% of cases (26). MPO-ANCA should be tested with antigen-specific immunoassays in any patient with eosinophilic asthma and clinical features suggestive of EGPA (such as constitutional symptoms, purpura, polyneuropathy, unexplained heart, gastrointestinal or renal disease, and/or pulmonary infiltrates or hemorrhage) (152), however, only approximately one-third of patients are ANCA-positive (9). Recently, a novel observation of ANCA reactivity in the sputum of seronegative EGPA patients was reported (63). ANCA reactivity was associated with more severe respiratory symptoms and sputum eosinophilia. It is now being investigated whether ANCA sputum could be useful as a diagnostic tool for serum ANCA patients with EGPA as well as to identify a subset of patients with eosinophilic asthma who are at increased risk of developing EGPA in the future (63).
Table 3. Selected studies investigated biomarkers in EGPA.
In EGPA, each organ may be affected; therefore, it is essential to conduct a thorough medical history interview and perform diagnostic tests assessing the functions and/or organ lesions. In addition, it is important to detect life-threatening organ involvement, as it requires rapid implementation of treatment (153). Generally, once EGPA is diagnosed, evaluating possible lung, heart, kidney, GI, and peripheral nerve involvement is recommended (153). Regarding the lungs and respiratory manifestations, a complete pulmonary diagnostic evaluation, comprising chest imaging at baseline and pulmonary function tests, should be performed (153). Every patient should have at least one chest radiograph, however, a CT scan is more sensitive and can provide a more precise assessment of lung lesions (153). Bronchoscopy with an evaluation of inflammatory cells in BALF can confirm pulmonary eosinophilia (defined as ≥25% eosinophils at differential cell count) (108). When DAH is present, BALF is bloodier and contains hemosiderin-laden macrophages (5).
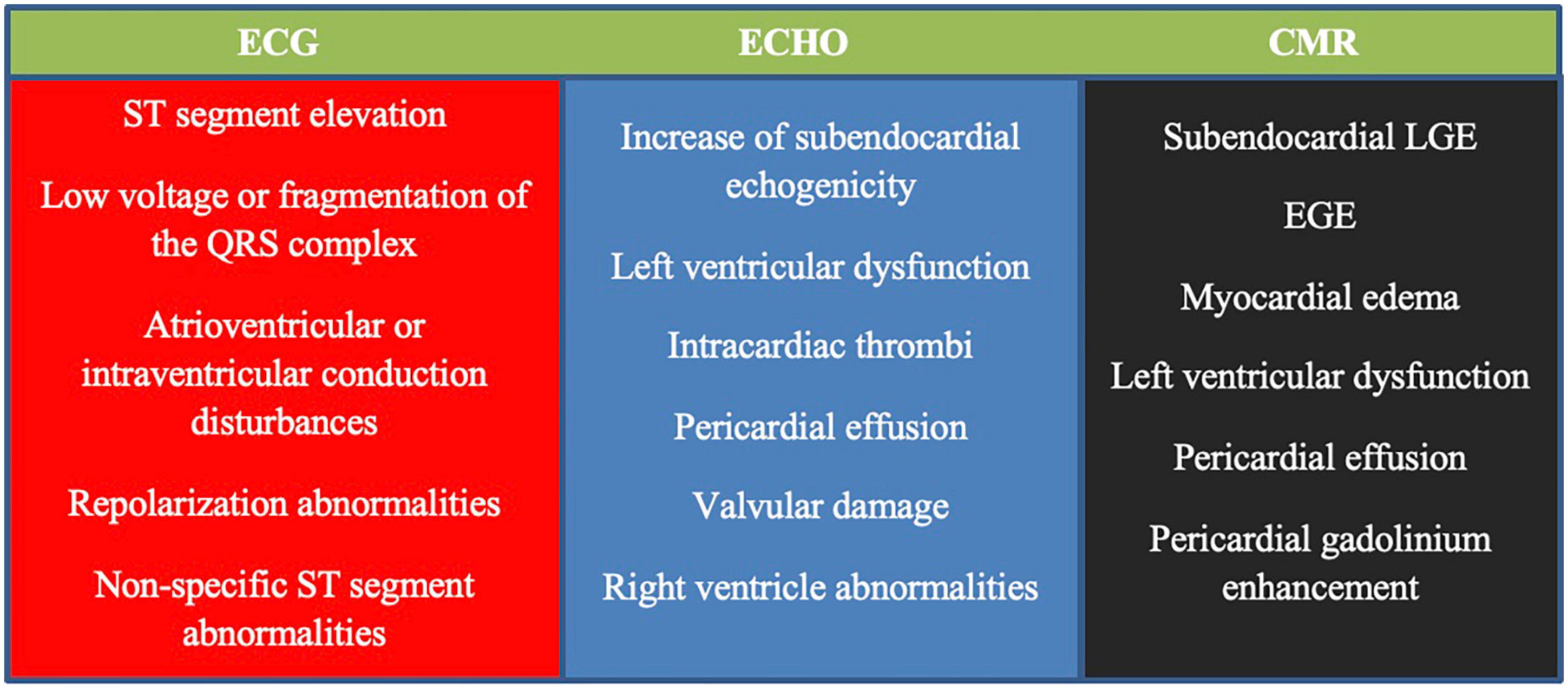
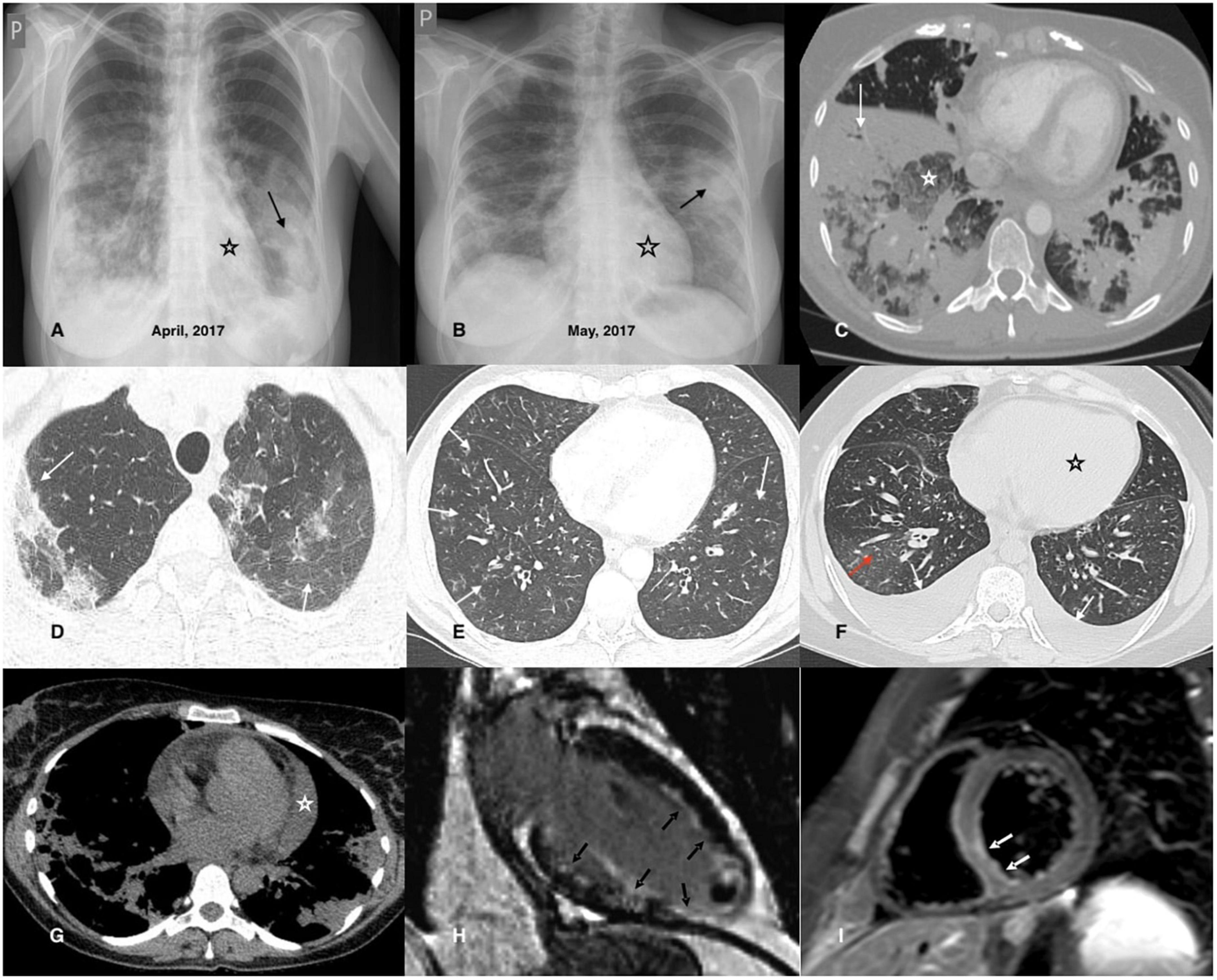
Cardiac involvement, in particular, is associated with poor prognosis (114, 154); therefore, basic cardiological examinations are recommended in all patients (at diagnosis and in case of relapse), irrespective of clinical symptoms (118, 153, 155). These examinations include resting electrocardiography (ECG), echocardiography (ECHO), and serum concentrations of brain natriuretic peptide (BNP) and troponin (118, 153, 155). The 24-h ECG monitoring can help detect arrhythmias that cannot be captured on resting ECG and may be life-threatening, leading to sudden death. Recently, cardiac magnetic resonance (CMR) imaging has been considered the gold standard technique for evaluating cardiomyopathies (118, 121, 156). It is a safe and non-invasive tool for the assessment of cardiac involvement in AAVs (118, 121, 122, 156–158). Furthermore, it can help identify the individual stages of myocarditis (with better visibility of endocavitary thrombosis) and determine the activity of the disease (121, 156, 158, 159), however, its particular diagnostic importance is in asymptomatic patients, in whom this manifestation can be easily overlooked (118, 120–123, 155, 158, 160, 161). CMR is also a useful tool for monitoring treatment efficacy and fibrosis (121, 161). Late gadolinium enhancement (LGE) by CMR (mostly of subendocardial location) is characterized by high sensitivity and specificity for the detection of cardiac inflammation and fibrosis (121), and its persistence following treatment has become a marker of cardiac disease severity (112). However, CMR abnormalities are detected in a high proportion of patients in clinical remission and their clinical and prognostic significance remains unclear (123, 161). Although endomyocardial biopsy (EMB) is still considered the gold standard for the diagnosis of myocarditis, it is not routinely performed due to the risk of complications and organizational difficulties. This procedure may be considered in doubtful cases, especially, when the diagnosis of EGPA has not been established (155). Signs of heart involvement in cardiological tests in EGPA are demonstrated in Figure 1. Figure 2 presents chest imaging findings in patients with EGPA.
Figure 1. Signs of heart involvement in cardiological tests in patients with eosinophilic granulomatosis with polyangiitis (EGPA) [based on Bond et al. (155)]. ECG, electrocardiogram; ECHO, echocardiogram; CMR, cardiac magnetic resonance; LGE, late gadolinium enhancement; EGE, early gadolinium enhancement.
Figure 2. Chest imaging findings in patients with EGPA. (A,B) Chest X-rays of a 42-year-old female patient diagnosed with EGPA. They demonstrate migrating patchy infiltrates with peripheral dominance (black arrow), characteristic for eosinophilic infiltrates, and rapidly enlarging heart related to its acute injury in the course of EGPA. (C) Chest CT axial image (lung window) of a 40-year-old female EGPA patient showing pre-dominant massive bilateral ill-defined areas of airspace (white arrow) and ground-glass (white asterisk) opacities located in both lower lobes of the lungs. (D) Chest CT axial image (lung window) of a 37-year-old female patient presenting pre-dominant areas of ground-glass opacities of varying intensity in both upper lobes of the lung (black arrows); histological examination of the transbronchial biopsy specimen reveals features of eosinophilic pneumonia and eosinophilic vasculitis. (E) The image refers to a 46-year-old male patient admitted for worsening asthma and eosinophilia. Chest CT axial image (lung window) shows a pre-dominant airway pattern-bronchi wall thickening and small centrilobular nodules (white arrows). BALF examination indicated pulmonary eosinophilia (65% of eosinophils), and the patient complained of numbness of the feet, and for several days purpura-type skin lesions occurred; MPO-ANCA was detected in the sera. The patient was diagnosed with EGPA. (F) Chest CT axial image (lung window) of a 34-year-old female patient with EGPA and cardiac involvement showing pre-dominant features of cardiac insufficiency; the ground-glass opacities with interlobular septal thickening (red arrow) corresponding to interstitial edema; bilateral pleural effusion (white arrow) and enlarged heart is also present (black asterisk). (G) Chest CT axial image (mediastinal window) of a 38-year-old female patient diagnosed with EGPA and cardiac involvement. In addition to bilateral pulmonary infiltrates, an enlarged heart and pericardial effusion is visible (white asterisk). (H) CMR refers to a 32-year-old male patient with EGPA; late gadolinium enhancement (LGE) image in vertical long axis cross-section showing subendocardial enhancement pattern (typical for EGPA) of the anterior wall, subendocardial and transmural enhancement of the inferior wall, inferior papillary muscle and the left ventricle (LV) apex (black arrows); thrombus seen as an unenhanced mass in the apical part of the LV cavity. (I) CMR refers to a 26-year-old male patient diagnosed with EGPA with cardiac involvement; a T2-weighted turbo spin-echo (TSE) image with fat saturation in the short axis mid-cavity cross-section, presenting edema in the infero-lateral segment of the LV (white arrows).
Renal involvement is the next poor prognostic factor; therefore, renal function tests and urinalysis should be performed in all cases at baseline and during follow-up (153). In asymptomatic patients, routine screening for GI and peripheral nerve involvement is not required, however, when symptoms are present, appropriate diagnostic procedures should be implemented (e.g., radiologic and/or endoscopic evaluation of the digestive tract in cases of gastrointestinal symptoms or electromyography and nerve conduction studies in cases suspected of nerve involvement). Other evaluations should be guided by clinical symptoms and physical examination (153).
In the presence of demonstrable lesions, biopsy procedures should be considered when feasible, and the patient’s condition allows it, however, histological examination is not strictly necessary (153). Although pathomorphological lesions are well-defined (necrotizing vasculitis, extravascular granulomas, and eosinophil infiltration of arterial walls and adherent tissue), it is extremely rare to find all of them simultaneously (<20% of patients) (135). The most commonly biopsied organs are the skin, nerves, and muscles. Although EGPA is considered a multi-organ disease, it is well known that limited forms may also occur. When a single extrapulmonary manifestation attributable to systemic disease is present, the disease may be called “formes frustes” of EGPA (108). In such situations, diagnosis is only possible by organ biopsy (162).
While EGPA share features with eosinophilic inflammation and vasculitis, the primary differential diagnoses include other eosinophil-related disorders and vasculitides. First, other common causes of secondary eosinophilia should be excluded from the study. Eosinophilia can be reactive to drugs, and severe reactions may result in organ manifestations mimicking EGPA (e.g., drug rash with eosinophilia and systemic symptoms, DRESS syndrome) (16). A careful history of medication use is crucial to emphasize the association between drug use and symptom onset. Second, helminthic infections need to be ruled out. Serology of Toxocara and Strongyloides stercoralis is especially recommended (153). Both are associated with high eosinophilia and can be clinically inapparent (163, 164). Other parasite investigations depend on the patient’s country of origin and travel history, however, stool culture, although it has low sensitivity, should also be performed (16). Next, screening for HIV should be performed, even though eosinophilia in this infection is usually mild (153). Lymphocytic variant reactive hypereosinophilia should also be considered, especially when skin manifestations dominate, with accompanying hypergammaglobulinemia. In such cases, lymphocyte immunophenotyping and T-cell receptor rearrangement analysis are indicated (153, 165).
Eosinophilic granulomatosis with polyangiitis often manifests as respiratory symptoms and lung infiltrates; therefore, it should be differentiated from eosinophilic lung disorders. ABPA and idiopathic EP share many features with EGPA, including eosinophilia, cough, dyspnea, and lung infiltrates. Moreover, a large proportion of patients with these diseases have asthma, which is a cardinal feature of EGPA (166, 167). ABPA is characterized by elevated serum Aspergillus fumigatus-specific IgE and IgG concentrations (149, 162) and often isolated fungal cultures in sputum or BALF (153, 166). However, distinguishing idiopathic EP from the second stage of EGPA remains challenging. The lack of organ symptoms and ANCA may help differentiate between the two (5), however, patients with idiopathic EP should be monitored for extrapulmonary symptoms because they may develop EGPA in the future.
Hypereosinophilic syndromes are the next most important consideration in the differential diagnosis of EGPA, given the overlapping clinical, radiologic, and histologic features, and biomarker profile (105, 168). Depending on the pathogenesis, three main types of HESs are distinguished: reactive (rHES), neoplastic (nHES), and idiopathic (iHES). In rHES, eosinophils are non-clonal and are thought to be driven by Th2 cytokines, mainly IL-5. This group includes patients with classified conditions associated with secondary eosinophilia, including EGPA (11, 165, 169) (however, eosinophilia in EGPA is not entirely secondary, as it has a partially genetic background related to the IRF1/IL5 gene variant) (56). In nHES, eosinophils are clonal and derived from eosinophil progenitors containing genetic alterations in oncogenic tyrosine kinase receptors, such as platelet-derived growth factor receptor A (PDGFRA) and B (PDGFRAB) and fibroblast growth factor receptor 1 (PGFR1) (105, 165, 169). This group also encompasses other myeloid neoplastic diseases with associated eosinophilia (with or without genetic abnormalities), as well as chronic eosinophilic leukemia. In turn, iHES is the largest type of HES (comprising about 50% of cases) and is a diagnosis of exclusion once reactive and neoplastic causes have been excluded (105, 165).
Although organ damage may be similar, some symptoms, such as hepatomegaly or splenomegaly, can be suggestive of clonal eosinophilia and nHES. In addition, a proportion of patients have abnormal peripheral blood counts, such as anemia (53%) or thrombocytopenia (31%), and patients with nHES usually do not respond to treatment with systemic CS (169). Screening for serum vitamin B12 and tryptase levels is sensitive to nHES and is recommended for all patients diagnosed with eosinophilia (153). In cases of suspected nHES, fusion gene testing is indicated. However, although only to be positive in nHES, a case of PDGFRA-positive EGPA has been described (42); therefore, some authors believe that testing for PDGFRA mutation should be performed routinely in all cases with hypereosinophilia, regardless of clinical manifestation, suspected EGPA, or ANCA-status (170).
Idiopathic HES is the most difficult to distinguish from EGPA, especially in ANCA-negative cases without vasculitic symptoms (165, 169, 171, 172). Both clinical and radiological symptoms are similar, but HES is usually not considered to have asthma or nasal polyps. However, this is not a distinguishing feature. A case series of iHES with the first presenting asthma-like symptoms has been recently described (173). In addition, it has been reported that approximately 10% of patients with HES have rhinitis (169). Histological examination also showed no differentiation. HES is typically characterized by tissue infiltration by eosinophils, which is also often found in cases of EGPA (105). Other findings, such as vasculitis and granulomas, are not typical for HES but are considered hallmark features of EGPA (2). Recently, among patients with a diagnosis of HES lacking asthma, a group characterized by necrotizing eosinophilic vasculitis confirmed by biopsy has been distinguished (174). The distinction of EGPA from this entity is challenging, especially because it cannot be excluded that both may be a part of a common spectrum.
There is a need for further research on suitable features for distinguishing EGPA from HES. Finally, a comparative study of 166 patients with blood eosinophilia (>1.000 cells/μL) and systemic manifestations demonstrated that CRP level was a sound diagnostic biomarker that could accurately differentiate between HES and EGPA, with low levels (<36 mg/L) suggestive of HES (175). Other authors have proposed a HES-suggesting laboratory index (HSLI) based on white and eosinophil blood count, with values ≥4.25 exhibiting a significantly high relative risk for HES (176). Recently, a scoring system (E-CASE) for differentiating EGPA from other types of eosinophilic disorders, including HES, has been proposed. It was based on the clustering analysis of 19 parameters of 58 patients with eosinophil-related diseases at a tertiary hospital and was extensively validated in 40 patients at another tertiary institution. This system includes clinical (peripheral nerve disorder, asthma, lung, and skin involvement), laboratory (RF positivity, MPO-ANCA positivity, IgE, and CRP elevation), and histological features (vasculitis detected by pathological examination), which have been awarded a point weight. A score ≥12 was considered positive for EGPA (177).
The next diseases that should be differentiated include other forms of vasculitis, especially AAVs. GPA and MPA share several clinical and histological features with EGP
留言 (0)