
記住我


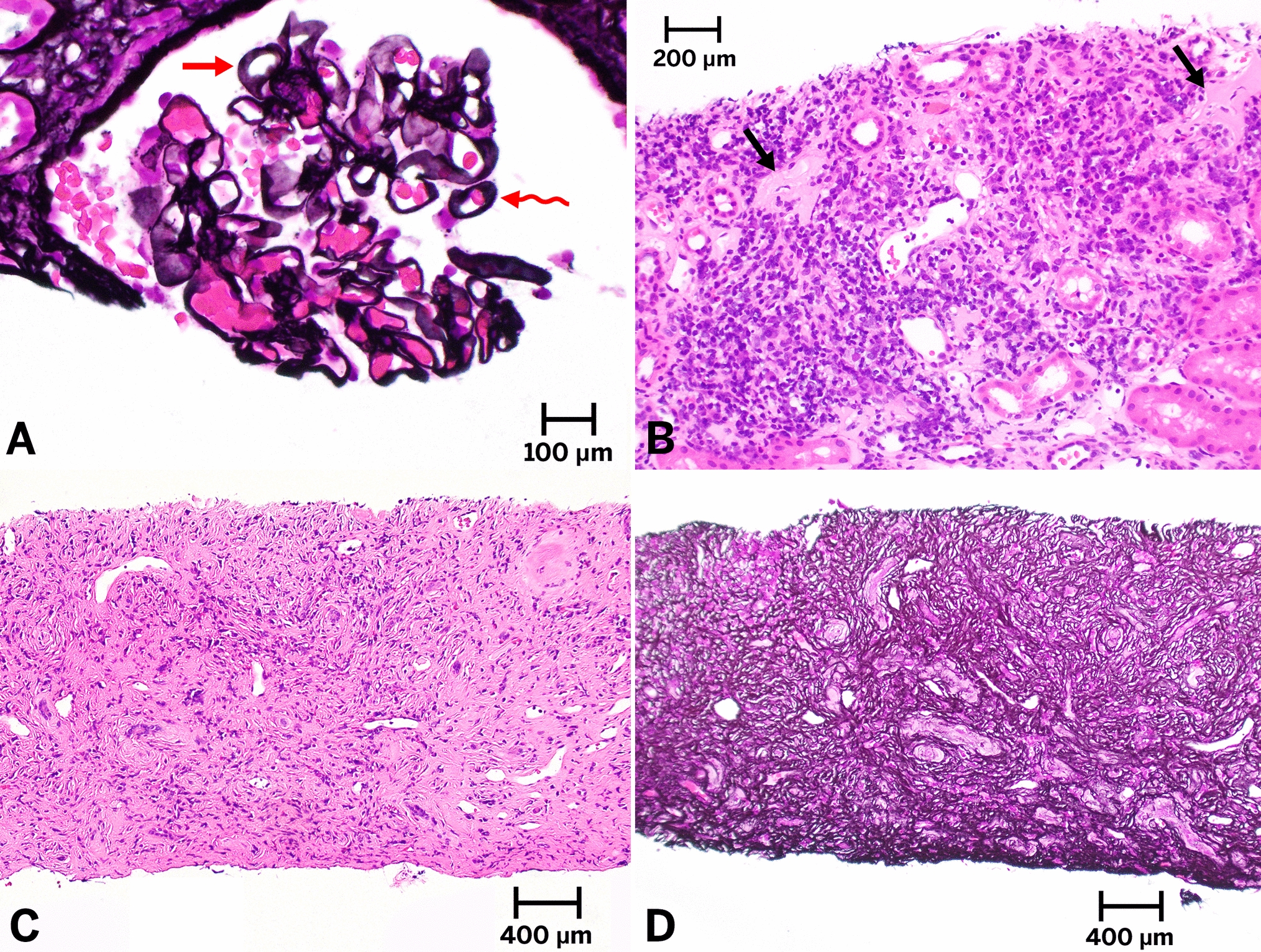
The etiology of pathophysiological mechanisms leading to muscle wasting is complex and includes mechanisms that increase muscular proteolysis through increased catabolism and apoptosis activation, and decrease the synthesis of muscle cells [11, 18]. Specific mechanisms, triggered by factors such as oxidative stress and inflammation [1, 11], include the caspase pathway, the lysosomal proteolytic system (cathepsin L), the calcium-dependent proteolytic calpain system [19], and not least the UPS, which are all strongly associated with PEW in CKD [2, 11] (Fig. 1). Through the insulin-like growth factor-1 (IGF-1)/phosphatidyl inositol-3 kinase (PI3K)/protein kinase B (Akt) pathway [20], one of the most explored anabolic signaling pathways affecting muscle in CKD, proteolysis in muscle cells occurs when there is a suppression of PI3-K activity in muscle that is induced, for example, by acidosis, leading to accelerated muscle proteolysis [20].
Fig. 1
Schematic representation of factors promoting NRF2-deficiency in patients with chronic kidney disease and mechanisms by which this may result in protein-energy wasting (PEW) and muscle wasting, by promoting oxidative stress/inflammation and activating the ubiquitin–proteasome system (UPS) pathway
Depletion of antioxidant enzymes (among others, superoxide dismutase [SOD1, SOD2, SOD3], hemoxygenase [HO1, HO2], glutathione peroxidase [GPx], catalase [CAT], and glutathione [GSH]) [21] promotes mitochondrial dysfunction and increases oxidative stress, causing damage to multiple cellular components such as DNA, proteins, and lipids, in metabolic and chronic diseases [15, 22]. In general, oxidative stress with increased levels of reactive oxygen species (ROS, superoxide [O2-], hydrogen peroxide [H2O2], hydroxyl radical [-OH] and peroxynitrite [ONOO-]) is the result of enzymatic activity of the mitochondrial respiratory system, such as cyclooxygenases (COXs), cytochrome P450, and myeloperoxidases [23]. In addition, alterations in the activity of various transcription factors regulating oxidant and antioxidant genes, such as Forkhead box O (FOXO), nuclear factor-kappa B (NFκB) and NRF2 have been studied in murine models but less so in humans [15, 24]. A systematic review of the relations between NRF2 and morbidity in CKD was recently published [16].
In response to oxidative attacks, cells turn on antioxidant defense systems, such as the NRF2 system, to maintain cell redox homeostasis and protect cells [22], while UPS, the main regulatory mechanism of skeletal muscle degradation, is activated. UPS involves three enzymes: ubiquitin-activating enzyme E1; ubiquitin-conjugating enzyme E2; and ubiquitin protein ligase E3 that regulates selectivity and specificity of protein degradation mechanisms [25]. Two muscle-specific E3 ubiquitin ligases, muscle RING finger-1 (MuRF1) and muscle atrophy F-box (MAFbx; atrogin-1) increase transcriptionally in skeletal muscle under atrophy-inducing conditions. MuRF1 participates in the contraction and structure of muscle proteins, while MAFbx participates in protein synthesis and muscle regeneration, but by acting together with myostatin it may also have a role in protein degradation leading to atrophy of skeletal muscle [25]. UPS could be considered responsible for the control and balance of both anabolism and catabolism of skeletal muscle proteins in conditions, such as prolonged fasting, diabetes and cancer, which are accompanied by high mRNA levels of MuRF1 and MAFbx, by activation of the NFkB system and repression of NRF2 [25]. However, the situation in CKD is unclear as information is rather scarce and mainly based on animal models.
Data from experimental studies suggest that intracellular activation of the caspase-12, 9 and 3 pathways stimulate the production of ROS and activate NFκB and the nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor, alpha [IkBα] [1, 26] to promote "ubiquitination" apoptosis and cellular autophagy; the latter prevents mitochondrial biogenesis at the muscular level by stimulating the degradation of cytosolic proteins (actomyosin) and organelles. These events are also highly regulated by the lysosomal pathway, in which the role of cathepsin L seems to be pivotal [2, 26]. Cathepsin L may also be present extracellularly, independent of the lysosomal fraction, playing a special role during atrophy [6, 27]. Also, calcium-dependent calpains (calpain 1, μ-calpain; calpain 2, m-calpain) are activated in hypoxic conditions [28], as well as in CKD associated with the induction of hypoxia factors 1-α, such as HIF1α, HIF2α, HIF3α which may contribute to muscle atrophy, muscle wasting and frailty [19] involving oxidation of contractile proteins, actin and myosin [26].
Furthermore, the COX pathway (especially COX-1 and COX-2) with synthesis of prostaglandins (PG, PGE2, PGF2α, PGI2 and PGD2) from arachidonic acid also regulates muscle regeneration and affects muscle degradation by modulating inflammation and myogenesis [29, 30]. COX-2/PGE2 responses induced by altered renal blood flow and pro-inflammatory cytokine activity contribute to the development of CKD, which associates with up to four-fold higher COX-2 than in absence of this disease, and to appetite loss and altered energy metabolism by blocking the central nervous system, thus promoting several pathways leading to PEW [31, 32]. Moreover, PGs derived from COX-2, but not from COX-1, are critical for muscle regeneration, which is consistent with their role in blocking repair of various systems and organs, including the kidneys [29, 30]. Notwithstanding, there are a limited number of studies exploring the role of NRF2 and UPS and the above mentioned factors for the associations of oxidative stress and inflammation with skeletal muscle alterations in CKD.
Similarly, the FOXO transcription factor is expressed in skeletal muscle in three main isoforms: FOXO1, FOXO3 and FOXO4. Evidence suggests that translocation of FOXO (especially FOXO1 and FOXO3) to the nucleus promotes increased expression of atrogin-1 (MAFbx) and MuRF1 (type E3 ligases) thus promoting muscle atrophy [1, 33]. Oxidative stress and inflammation [26] are regulators of the expression of MuRF1 and MAFbx via p38 mitogen activated protein kinase (MAPK), FOXO and NFkB [25].
The central importance of NFkB is highlighted by the fact that it regulates the expression of many genes including those responsible for muscle proteolysis. Activation of NFkB via the UPS and the MuRF1 pathways induces muscle degradation and muscle wasting, which in turn induces cellular apoptosis [19]. Higher levels of NFkB have been observed in hemodialysis patients with poor nutritional status, reinforcing the hypothesis that inflammation is a key driver of PEW [34]. Furthermore, blocking the translocation to the nucleus by pharmacological or genetic inhibition of NFkB prevents the expression of several components of the proteolytic machinery including MuRF1, muscle synthesis and preservation [6]. NRF2 is also influenced by the inflammatory cytokine tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), and as NRF2 activation inhibits TWEAK-induced atrophy in myotubes, NRF2 may protect skeletal muscle from TWEAK-induced cell death [35].
NRF2 protein (605 amino acids, belongs to a subset of the basic leucine zipper family proteins) is responsible for antioxidant transcription (as a master regulator) and, under normal conditions, it is bound to KEAP1 [36, 37]. This ubiquitin conjugation, NRF2-KEAP1, favors rapid proteasome degradation in the cytoplasm by activating the ubiquitin ligase complex Cul3-E3; however, in a state of stress, NRF2 is released from KEAP1 and rapidly accumulates in the nucleus, activating the antioxidant response element (ARE) in the promoter region of many antioxidant genes [22], which in turn leads to increased regulation of antioxidants and phase II detoxifying enzymes [36]. Nuclear respiratory factor 1 (NRF1) contains multiple RNAs in the promoter part of its gene, which are necessary to promote NRF2 activity from the induction of ROS [38]. Thus, NRF2 is sensitive to redox state and plays a role in the regulation of UPS components [39]; therefore, it modulates pro-apoptotic signals, such as NFkB, ASK1, BAD, BAX, AIF, AP1, peroxisome proliferator-activated coactivator gamma 1-alpha (PGC1α) and caspases 9 and 3, but the response depends on the accumulation of ROS and depletion of GSH [18].
The effect on skeletal muscle remains poorly understood; whereas higher NRF2 expression has been observed in non-dialysis CKD patients, hemodialysis patients exhibited reduced NRF2 gene expression, which was associated with increased NFkB gene expression possibly related to systemic inflammation. Considering that the uremic milieu in CKD patients (CKD stage 4) has been associated with up-regulation of NRF2 [40], it is of interest that a recent investigation evaluating the humanin peptide and the mitochondrial open reading frame of 12S rRNA-c (MOTS-c) related to cell survival, suppression of apoptosis in oxidative stress or starvation, as well as enhanced insulin secretion and action found that patients with stage 5 CKD had increased circulating levels but reduced local muscle expression of humanin [41]. On the other hand, in CKD stage 5 patients, MOTS-c levels were observed to be reduced in both serum and muscle, together with a reduction of NRF2 expression in muscle [41]. Protein degradation in skeletal muscle has been scarcely described in relation to this pathology [42]. However, a study in older adults—presumably with some degree of aging-related sarcopenia—reported lower expression of NRF2 [43]. This might affect redox homeostasis and alter skeletal muscle structure and function through altering the balance between oxidizing and antioxidant agents [18, 44].
While clinical and experimental data suggest that hyperphosphatemia, a prominent alteration in CKD, accelerates muscle wasting, the underlying mechanism remains unclear. However, data in mice suggest that hyperphosphatemia suppresses myogenic differentiation in vitro and promotes muscle atrophy in vivo through oxidative stress-mediated protein degradation and both canonical (ROS-mediated) and non-canonical (p62-mediated) activation of NRF2 signaling [45].
In general, myocytes in skeletal muscle are enriched with mitochondria, that could account for as much as 1/3 of the total weight of the cell, facilitating the excessive production and accumulation of ROS, dependent on CKD stage and age [15, 18, 44]. NRF2 deficiency may exacerbate age-related mitochondrial oxidative stress in aged skeletal muscle [46] and be part of an intermediate inflammatory phenotype that promotes burden of lifestyle diseases that accumulate with age [47]. The decrease in the number of fat-free myocytes and the low transcription of NRF2 [44] may be related to several factors, such as inadequate diet, hyperparathyroidism, depression, dementia, osteoporosis, periodontitis [43], dialysis, uremic toxins [48], obesity, hyperglycemia, variants of NFE2L2 gene (encoding NRF2 protein) [22, 49], metabolic acidosis and endothelial dysfunction [23, 34]. All these factors promote muscle degradation and may contribute to skeletal muscle dysfunction by inducing ubiquitination, lipid peroxidation and activation of apoptotic processes and autophagy with mediators such as calpain and caspases [18]. Moreover, although little is described in humans, the catalyzed reaction of COX appears to contribute to PEW through the NRF2 signaling pathway [14, 18]. The NRF2-related molecular mechanisms leading to muscle dysfunction have not been fully described; new lines of study are open on how NRF2 dysregulation affects muscle mass, quality and function, and leads to PEW in the context of CKD [50].
A better understanding of these processes is of importance to design therapeutic strategies to reverse these complications. To date, there is, with few exceptions, a lack of studies clinically demonstrating the safety and effectiveness of drugs targeting NRF2 to address these complications in humans [51]. A clinical trial in patients with obesity, diabetes, and stage 4 CKD who received bardoxolone methyl demonstrated improved glycemic control, weight loss, decreased lipid accumulation, and reduced inflammation through activation of NRF2 and inhibition of NFkB [40]. On the other hand, a growing number of studies have explored non-pharmacological approaches to enhance NRF2-related mechanisms affecting protein synthesis and degradation to prevent muscle proteolysis or promote muscle synthesis, including resistance training [38]; however, in a study conducted in NRF2-deficient mice during endurance exercise stress, it was proposed that other NRF2-independent mechanisms such as PGC1α are activated by inducing mitochondrial biogenesis in aging skeletal muscle cells [18], which may also be activated by derivatives or food supplements known as nutraceuticals [14, 52].
留言 (0)